.png)

The human body is a mosaic of cells. Immune cells ramp up inflammation to fight infections; stem cells differentiate into diverse tissues; cancer cells evade regulatory signals to divide uncontrollably. Despite their remarkable differences, however, each human cell carries (nearly) the same genome. A cell’s distinctiveness arises not just from differing DNA, but rather in how each cell uses that DNA.
In other words, a cell’s properties emerge from variations in gene expression, or the switching of genes “on” and “off” across time. A cell’s gene expression patterns—expressed in terms of RNA molecules, which are themselves transcribed from the genome—determine not only its cell type but also its cellular state: changes in a cell’s gene expression can reveal how it moves from healthy to inflamed to cancerous. By measuring the RNA transcripts within cells with or without a chemical or genetic perturbation, it is possible to train AI models capable of predicting how a cell’s gene expression patterns—a key driver of the cell’s “state”—will change. Such models could even predict responses to perturbations that the model hasn't encountered before.
About 90 percent of drugs fail clinical trials due to poor efficacy or unintended side effects. Each drug that researchers test in the laboratory, or in a patient, is essentially a tailored probe designed to perturb cells in a particular way. A highly predictive virtual cell model could therefore help researchers discover new drugs capable of shifting cells between states—from “diseased” to “healthy”—with fewer off-target effects to boost clinical success rates.
Introducing State
Today, Arc is releasing its first generation virtual cell model, called State. The model is designed to predict how various stem cells, cancer cells, and immune cells respond to drugs, cytokines, or genetic perturbations. State is trained on observational data from nearly 170 million cells and perturbational data from over 100 million cells across 70 cell lines, including data from the Arc Virtual Cell Atlas. The model is available for noncommercial use. More details can be found in the preprint and GitHub repository.
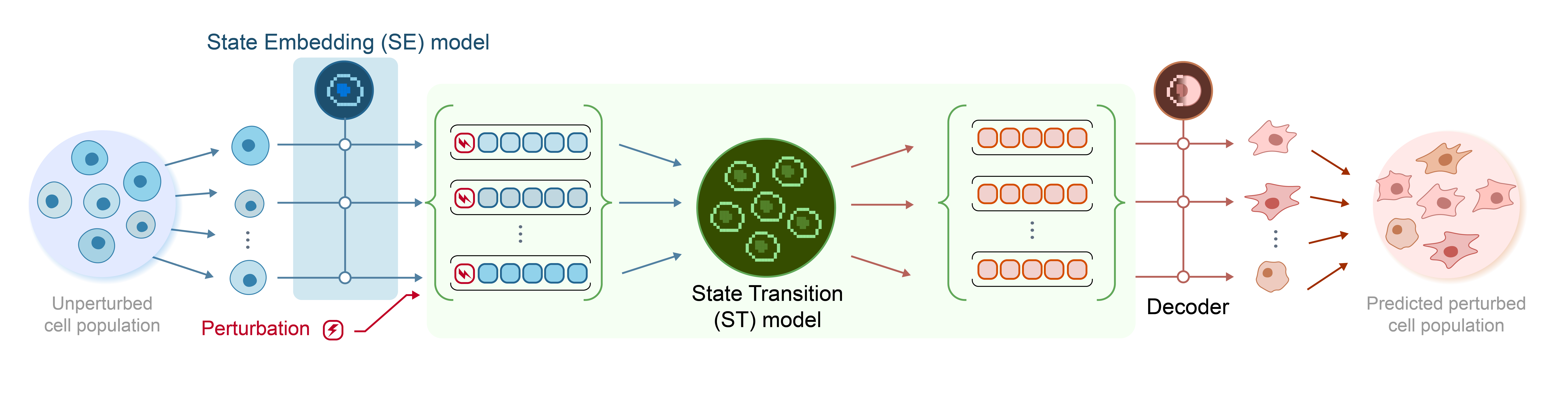
Using State is simple: given a starting transcriptome and a perturbation, State predicts the resulting shifts in RNA expression. State is made from two interlocking modules, called the State Embedding (SE) model and the State Transition (ST) model. The optional SE model converts transcriptome data into a smooth multidimensional vector space that computers can more easily understand and is more invariant to technical noise. Cells of the same type, such as leukemia cells or neurons, cluster together in this vector space. The ST model predicts how cells will transition between different parts of the learned manifold in response to a given perturbation. This model is built on a bidirectional transformer architecture that uniquely leverages self-attention over sets of cells, such that ST can flexibly capture biological and technical heterogeneity (such as cell cycle state or biases in RNA-seq data) without relying on explicit distributional assumptions.
State is trained on single-cell perturbation data from more than 100 million cells (Tahoe-100M, Parse-PMBC, Replogle-Nadig), more than any other model to date. It significantly outperforms existing, state-of-the-art computational approaches to predict how transcriptomes change after perturbations in new cell contexts. During benchmarking on Tahoe-100M, State demonstrated a 50 percent improvement in distinguishing perturbation effects and achieved twice the accuracy in identifying true differentially expressed genes compared to these existing models. To the best of our knowledge, State is also the first model to consistently beat simple linear baselines.
Why perturbation data?
State is initially focused on modeling single-cell RNA sequencing data because it is currently the only unbiased single-cell resolution data that researchers can generate at large scale with reasonable cost. Unfortunately, sequencing data is usually purely observational and thus generally insufficient for inferring causal relationships in cell biology. Even with observational data from millions of cells, a virtual cell model cannot zero in on causal effects from which observed correlations emerge. Learning causality is essential for building a true "virtual cell" model grounded in biological mechanisms.
We are compensating for this data deficiency by collecting large-scale perturbation data: namely, data generated experimentally (e.g. with CRISPR tools) where specific genes are deliberately altered to observe their effects on the cell. Unlike observational data, perturbative data captures causal relationships between genes, directly reflecting the underlying biological mechanisms. Whereas it might take tens of thousands of observations to infer a direct relationship between two genes, perturbative data can capture the same interaction with a single measurement. At Arc, we’re integrating technology development with machine learning in a unique way, allowing us to scale data collection rapidly and innovate in modeling approaches.
To date, most single-cell data comes from small studies where technical and source batching degrades our ability to seamlessly integrate data across many projects. At Arc, we developed and launched scBaseCount, the first agentic AI in this space tasked with uniformly collecting and analyzing single cell data to minimize analytical artifacts. scBaseCount is currently the largest open-source repository of single cell data. State itself is also able to model these types of “confounding” factors directly, which enables it to integrate a large number of distinct datasets from different labs around the world.
Even though State is just the first version in what we hope will ultimately be a string of steadily improving models. As training data for the virtual cell grows, so too does its predictive accuracy. This may seem like an obvious outcome, given that scaling laws have been observed in other domains for several years, but this has only recently been established for biology. Last year, we revealed scaling laws for language modeling of DNA for the first time.
Looking Ahead
Use cases for State may follow similar patterns to protein-folding models. AlphaFold became useful not only because it could accurately predict protein structures, but also because researchers found ways to integrate its predictions into workflows. By quickly predicting protein structures, for example, scientists could also more quickly discover small molecules likely to bind to those proteins.
Similarly, researchers can use State and future models not only to simulate how cells respond to perturbations, but to then use those predictions to nominate and discover new drugs experimentally.
The ultimate reason to make a virtual cell model, though, is to help scientists explore a much larger space of combinatorial possibilities. Any living cell can be altered in a vast number of ways, and there is no way to test every genetic mutation or drug treatment that might treat, say, a cancer cell. A highly predictive virtual cell model will address this issue. State is a first step in this direction, and our goal is to eventually match experimental precision with future versions of our virtual cell models. This will enable scientists to run millions of in silico perturbations to “narrow down” their hypotheses in the process of making original discoveries.
To help with this, we have also unveiled Cell_Eval, a comprehensive evaluation framework for virtual cell modeling that advances beyond conventional metrics in the field, such as those based on expression counts, to include a suite of biologically relevant and interpretable metrics focused on differential expression prediction and estimation of perturbation strength. We hope that Cell_Eval will assist in the transparent assessment of current and future generations of virtual cell models, much like LMArena has played a leading role in comparing LLM developments in text, image, or vision modeling.
We’re unveiling this first State model in the hopes that biologists will use it and begin devising ways to incorporate it into their own work. We welcome all feedback as we work to make this model maximally useful to the research community.

![The 8pen input method for phones (2010) [video]](https://www.youtube.com/img/desktop/supported_browsers/edgium.png)

