.png)

Introduction
The ability to encode genetic information while executing a diverse range of functions has allowed RNA to gain traction as a powerful molecular platform for the development of therapeutics, diagnostics, and synthetic biological systems1,2,3,4,5,6,7,8,9,10,11,12. The quartet of canonical nucleotides and simple rules of Watson-Crick base pairing make the design of specific RNA molecules a far more computationally tractable problem than analogous protein-based systems. Extensive work has been conducted to develop robust tools for the prediction of RNA secondary structure and structure-based inverse design13,14,15,16,17,18,19,20,21,22. However, there does not exist an equally generalized set of tools for computational engineering of RNA function. As RNA-based systems have become more sophisticated and widely used, a disconnect between deterministic thermodynamic properties and resulting RNA function has grown, often necessitating experimental screening of many RNA sequences3,5,7,9. Accordingly, data-driven methods that can decipher the relationship between RNA sequence and function are essential for expediting RNA device design and improving their performance in biomedical applications.
To facilitate data-based approaches to RNA engineering, massively parallel reporting assays have been designed across a variety of contexts, resulting in a number of datasets mapping functional RNAs to their experimental performance23,24,25,26,27,28,29,30,31,32,33,34. Toehold switches35,36, 5′ untranslated regions (UTRs)37, CRISPR guide RNAs38, and ribosome binding sites (RBSs)39 are all examples of RNA sequences that have been functionally characterized using high-throughput screening. In each of these cases, predictive models that operate on a one-hot-encoded representation of the input sequences have been constructed alongside the datasets. The architectures and performance of these models vary widely, despite their common task of predicting RNA function. This has resulted in several available predictive methods that are tuned extensively to their specific functional task but cannot be applied to the full span of settings in which RNA has found use. Just as thermodynamic algorithms for sequence-structure relationships can be applied regardless of the class of RNA being analyzed, we aimed to develop a similarly generalized framework for predicting RNA sequence-function relationships and designing new instances of RNA molecules with a pre-determined function. A robust set of tools that enable the prediction of RNA function and the inverse design of functional sequences could greatly accelerate the deployment of RNA systems in both research and industrial settings.
Despite both sequence and structure playing a large role in the function of RNA molecules, there currently does not exist a generalized deep learning framework that incorporates both properties to inform predictions. Here we report a deep learning approach that incorporates the sequence and structure of RNA molecules (SANDSTORM) to predict the function of diverse classes of RNAs. Using SANDSTORM neural networks, we predict the functional activity of 5′ UTRs, RBSs, CRISPR guide RNAs, and toehold switch riboregulators. By combining these predictive models with generative adversarial RNA design networks (GARDN), we generate novel and realistic instances of several of these RNA classes that also exhibit desirable functional properties. This joint design method demonstrates that the abstractions of secondary structure learned by the SANDSTORM architecture can be effectively leveraged by the generator during training, resulting in generated constructs that reflect experimentally characterized thermodynamic properties. The ability to both predict and generate RNA sequences with tailored functional activity across a range of heterogenous application domains could serve as the foundation of a wide range of next-generation computational RNA tools.
Results
Implementing an efficient representation of RNA secondary structure
In order to predict function using both sequence and structure, we first developed a representation of RNA secondary structure that could be supplied to a predictive model in parallel with one-hot-encoded sequences (Fig. 1, Supplementary Fig. 1). To do so, we took inspiration from the field of image classification, where manually engineered data features have been surpassed by deep convolutional models which learn their own abstractions of input data40. By constructing a basic array that encoded only the locations of possible base pairing interactions, we hypothesized that a deep convolutional neural network (CNN) would be able to construct similar abstractions related to the structure of an RNA molecule during the training process. The novel structural array we present (Supplementary Fig. 2) is thematically similar to others that have been developed for RNA inverse design15,19,35, but offers the time efficiency necessary to be practically useful in a deep learning pipeline while avoiding any assumptions inherent to classic structural prediction algorithms (Supplementary Fig. 3).

a SANDSTORM expands upon previous sequence-to-function neural networks by incorporating both sequence and secondary structure array input channels. These paired inputs are passed through parallel convolutional stacks that form an ensemble prediction of input RNA function (see Supplementary Fig. 1a for a detailed depiction of SANDSTORM). b GARDN is a generative adversarial network architecture which accepts a random variable input (Z) and is tasked with designing realistic examples of functional RNAs (see Supplementary Fig. 1b for a detailed depiction of the GARDN generator). c A trained GARDN generator can be paired with a SANDSTORM predictive model to return realistic sequences with targeted experimental values.
Learning interpretable structural information with a simulated dataset
We evaluated a CNN’s ability to convert our structural array into interpretable structural information using a simulated dataset of toehold switches and several decoy sequences (Fig. 2a). Toehold switches are riboregulators that modulate translation of an output protein in response to a complementary RNA input molecule3. These devices are designed to hide the RBS and start codon from the translational machinery by placing them within a cis-repressing stem. This stem can be unwound by an RNA trigger that is complementary to the 5′ end of the switch, releasing both the RBS and start codon. Because these devices require both sequence and structural motifs to function, they serve as an ideal candidate to demonstrate the performance improvement realized by using both properties as inputs. A dataset was constructed consisting of canonical toehold switches and four types of decoy sequences: sequences containing only an RBS motif and random nucleotides; sequences containing only a start codon motif; sequences containing both a start codon and RBS, but do not fold into the toehold switch structure; and sequences that fold into the toehold switch structure, but do not contain the necessary sequence-level motifs.

a A dataset of toehold switches and several types of decoy sequences was utilized to determine whether a CNN could differentiate sequences on a structural basis. Sequences consisted of canonical toehold switches, decoys containing only an RBS motif (RBS decoys), decoys containing only a start codon motif (AUG decoys), decoys containing both an RBS and start codon motif (RBS + AUG decoys), and decoys that adopted the canonical secondary structure but did not contain the necessary sequence motifs (binding decoys). b A CNN trained only on one-hot-encoded sequences was not able to perfectly classify the canonical toehold switches from the RBS + AUG decoys, which are only differentiable at a structural level. c The SANDSTORM CNN accepting paired sequence and structure arrays was able to identify both the sequence and structural features required to classify the simulated dataset. Source data are provided as a Source Data file.
We compared the performance of a model accepting only one-hot-encoded sequences as inputs to the SANDSTORM model accepting a paired sequence-structure input at the task of classifying each member of this simulated dataset (Fig. 2b, c). As expected, the one-hot-encoding model did not perform as well as the dual-input model when differentiating between canonical toehold switches and decoys that contained the necessary sequence motifs but did not fold into the correct secondary structure (single-input sequence-only model AUC = 0.72, SANDSTORM model AUC = 0.97). As these two categories are only differentiable at the secondary structure level, we concluded that our novel array enabled the model to learn useful abstractions of RNA structure to inform predictions.
To further validate that the paired-input classification model was identifying meaningful representations of secondary structure, we applied the integrated gradients technique41 to the trained predictive model along the structural input channel (Supplementary Fig. 4a, b). When examining a canonical toehold switch, this approach reveals a strong model preference for base pairing interactions in the region of the stem and a strong aversion for proximal off-target base pairing interactions. This agrees closely with the minimum free energy predicted structure of the sequence, indicating the model’s ability to correctly identify and leverage interpretable structural motifs. As structural features are products of primary sequence relationships, we further hypothesized that a sufficiently complex sequence-only model would be able to eventually approach the performance of the dual-input classifier. We explored this question by increasing the size of the receptive field for the sequence-only model and by increasing the number of trainable hyperparameters (Supplementary Fig. 5), both of which should allow the sequence-only model to construct more complex abstractions of the inputs it receives. In agreement with this hypothesis, we found a steady increase in the sequence-only model’s ability to correctly classify the structure-dependent decoy class as we increased the size of the receptive field and total number of trainable hyperparameters. Despite these improvements for the sequence-only model, the dual-input method demonstrated consistently better performance across parameter variations (Supplementary Fig. 5).
SANDSTORM models predict the function of multiple classes of RNAs
After demonstrating the capability of the SANDSTORM architecture to account for secondary structure in a more efficient manner than a sequence-only comparator, we sought to determine if this approach could be generalized to predict the function of different classes of real RNA molecules (Fig. 3). As our goal was to develop a generalized framework for predicting RNA function, we designed a single, efficient SANDSTORM CNN architecture that could be applied to many RNA functional prediction tasks without sacrificing performance. Our model consisted of a sequence input channel and a structure input channel, each connected to an independent stack of convolutional layers. The outputs of the convolutional channels were concatenated before being passed through a final series of densely connected layers to allow the model to learn complex relationships between the features abstracted from each input. The model consisted of as few trainable parameters as possible and the hyperparameters were not tuned for each presented test case. The mean squared error (MSE), R2, and Spearman correlation for SANDSTORM and the previously published model for each regression task are compared, averaging across three randomized training-testing splits (Supplementary Data 1 for datasets).

a SANDSTORM prediction metrics for toehold switch ON and OFF values compared to NuSpeak/STORM. b SANDSTORM predictions for 5′ UTR (untranslated region) mean ribosome loading compared to Optimus 5-prime. c SANDSTORM model predicting RBS (ribosome binding site) translation efficiency compared to SAPIENS. d SANDSTORM predictions of Cas13a collateral cleavage efficiency using guide RNA-target pairs compared to ADAPT. Bars represent means across three independent training-testing splits ± s.d. Links to comparator datasets and code repositories are available in Supplementary Data 1. Source data are provided as a Source Data file.
Toehold switch prediction
Angenent-Mari et al.35 conducted an assay that sorted a large library of toehold switches based on their resulting fluorescence levels measured in both the native folding conformation, which corresponds to the OFF state, and when fused to the cognate trigger, which is equivalent to the ON state. This dataset therefore consists of toehold switch sequences that are mapped to a fluorescence readout in the ON and OFF states, with desirable sequences having the highest ON/OFF ratio possible. Despite the wealth of data mapping sequence to function for toehold switches, there is currently no theoretical framework that can reliably link sequence or thermodynamic characteristics to ON/OFF ratios. Furthermore, previous studies attempting to use engineered thermodynamic features as the inputs to a multi-layer-perceptron similarly failed to produce strong predictions35. The SANDSTORM-toehold architecture can match the most successful model from previous studies35,36 (MSE p-value = 0.13, two-sided t-test) values while using 67% fewer trainable parameters (Fig. 3a).
5′ UTR prediction
Several cis-regulatory elements affect the translational efficiency of full-length messenger RNAs in eukaryotic cells. One such element that can have a profound effect on protein production is the 5′ UTR; however, a detailed prediction of the regulatory effect cannot be captured by first-order sequence or structural relationships42,43. To survey the space of regulatory UTRs, Sample et al.37 conducted a high-throughput polysome profiling assay and constructed a predictive CNN using the resulting sequence-function dataset. Our SANDSTORM-UTR architecture was able to match the previously reported performance metrics (MSE p-value = 0.32, two-sided t-test) on this dataset (Fig. 3b) while using only 9.7% of the number of trainable parameters, suggesting that the predictive capabilities of SANDSTORM models can be applied across RNA classes.
RBS-flanking sequence prediction
The RBS sequence motif is required for translation initiation in prokaryotic systems. While the core motif is necessary for translation to occur, the variable nucleotides surrounding the RBS can have a large impact on the resulting degree of protein production11,44. This effect was quantified in a high-throughput manner by promoting translation of a site-specific DNA recombinase off a library of different RBS-flanking sequences39. When translated, this recombinase can flip the orientation of downstream section of the DNA molecule, allowing the fraction of DNA readouts that are flipped after sequencing to serve as a proxy for translation efficiency. As with the other cases reported here, the relationship between RBS-flanking sequences and translation efficiency was originally modelled using a sequence-only probabilistic deep learning framework. The SANDSTORM-RBS model was able to again match the predictive performance of the previously published approach (R2 p-value = 0.50, Spearman p-value = 0.95, two-sided t-test) while using 95% fewer trainable parameters (Fig. 3c). As the original model published with this dataset used a probabilistic loss instead of MSE, the performance comparison consists of only the correlations between predicted and ground-truth outputs.
CRISPR Cas13a target-guide prediction
CRISPR/Cas nucleases enable site-specific cleavage of nucleic acids based on the spacer sequence supplied by a guide RNA2. Predicting the efficiency of on- and off-target sequences that could be cleaved by Cas nucleases is a major challenge in the development of CRISPR systems and has been approached with a variety of different methods. To tackle this challenge, high-throughput methods31,38 have been developed to map the relationship between gRNA/target RNA pairs and resulting Cas13a collateral cleavage efficiency. The SANDSTORM-Cas model is capable of significantly improving on the performance of the previously characterized sequence-only model38 (MSE p-value = 2.5 × 10−8, two-sided t-test) while using only 42% of the trainable parameters (Fig. 3d). Additionally, this test case demonstrates that SANDSTORM architectures are capable of predicting RNA functions that are not tied directly to a translational readout, further supporting the generalizability of this approach.
Implementing a generative architecture for RNA design
After establishing a generalized approach to predict RNA function, we aimed to develop a similarly robust framework for designing functional RNA sequences. While generative modelling has recently gained traction across a number of different molecular design tasks45,46,47,48,49,50,51,52,53 the heterogeneous functions of RNA create a distinct set of design constraints that are not easily accounted for in a generalized manner. Certain functional RNA molecules, such as 5′ UTR sequences, do not require the inclusion of specific sequence or structural motifs in order to function, while many naturally occurring RNA sequences must strictly adhere to a target structure or incorporate regulatory motifs to demonstrate their intended effect. More complex still are synthetic RNA switches, which must adopt a target conformation while retaining nucleotide diversity within their programmable target recognition regions. Various generative and thermodynamic design tools have flourished individually in each of these domains9,10,11,13,14,15,18,22,23,24,26,28,36,37,38,39,45,47,49,50,51,52,53; however, a generative counterpart to our SANDSTORM architecture that can be used regardless of the sequential or structural constraints of the design task has yet to be developed for functional RNA molecules.
In order to unify the strengths of both thermodynamic- and deep learning-based design approaches, we developed Generative Adversarial RNA Design Network (GARDN) models that could incorporate both variable and conserved sequence motifs, experimental predictions, and adherence to a target secondary structure. Generative Adversarial Networks54,55 have demonstrated striking results across a number of different domains, and further offer the ability to be directly optimized by a trained predictive model45. As a proof of concept for utilizing a generative adversarial network in tandem with our structure-informed predictive tools, we began with the least-constrained task of generating 5′ UTR sequences. While these sequences can have a profound effect on translation levels, there are no specific requirements that a design algorithm must adhere to for a candidate UTR to be considered valid. We first characterized a GARDN model that could faithfully recreate the sequential and structural attributes of the entire dataset used during training (Appendix 2, Supplementary Fig. 6). When optimizing this model with our trained SANDSTORM-UTR predictor, we could furthermore fine-tune the generated outputs to recreate the distribution of lowest- or highest-expression sequences while reflecting both global and local structural profiles (Appendix 2, Supplementary Fig. 7).
Following this proof of concept, we investigated the utility of the GARDN approach on the more specific design task presented by RBSs. These sequences must incorporate some portion of the Shine-Dalgarno sequence (AGGAGG) to efficiently promote translation; however, the context surrounding the core motif can greatly impact the resulting protein expression. We trained a GARDN model on the RBS dataset and optimized the output sequences using a trained SANDSTORM-RBS predictor. The resulting post-optimization sequence group favored the incorporation of the core Shine-Dalgarno sequence compared to the non-optimized sequences returned by the GARDN model (Fig. 4a, top, middle), indicating that sequence-function relationships learned by the predictor can directly guide the sequences returned by the generator during optimization. This preference was in line with the three highest-performing sequences reported in the experimental dataset (Fig. 4a, bottom), which also demonstrate the inclusion of this motif. We experimentally validated the performance of 5 RBS sequence pairs optimized by the GARDN-SANDSTORM routine, demonstrating a geometric mean fold change of 28.2 between pre- and post-optimized sequences (Fig. 4b). Strikingly, 3 out of 5 of the GARDN-SANDSTORM optimized sequences also exceeded the translational activity of the three highest performing sequences reported in the original dataset used for training (Fig. 4b). Adherence to the constraints of RBS design while also accounting for experimental performance highlights the utility of the GARDN-SANDSTORM across diverse RNA settings.

a The nucleotide distributions of RBS (Ribosome-binding site) sequences returned by the GARDN model (top), the GARDN model optimized by a trained SANDSTORM-RBS predictor (middle) and the three highest performing sequences from the experimental dataset used for training (bottom) (see Supplementary Data 2 for sequences). b GFP expression measurements in BL 21 Star (DE3) E. coli as determined by flow cytometry for RBS sequences designed using the GARDN-SANDSTORM approach and the three highest-performing sequences reported in the original dataset used for training. See Methods (Flow Cytometry Analysis) and Supplemental Information for representative gating strategy. Bars represent the fluorescence measurement mean ± s.d. Post-optimized bars denoted with * indicate a statistically significant (p < 0.05) increase in fluorescence compared to all three high-throughput examples as measured by a one-sided t test. Source data are provided as a Source Data file.
While the GARDN-RBS designs show significant performance improvements, the specificity of this RNA design task represents a challenge that generative models are intrinsically well-suited to address. Notably, when a highly conserved sequence motif is predominantly driving RNA function, a generative model must primarily identify and reproduce those nucleotides which are conserved across active members of the sequence group in order to return a valid candidate. We aimed to expand this framework to the far more challenging task proposed by the design of RNA switches, in which candidate sequences must adhere to a target secondary structure in a sequence-independent manner while also incorporating conserved nucleotide domains. For this task, we focused on the design of toehold switch riboregulators.
During preliminary tests, we found the generative architectures that enabled UTR and RBS generation struggled to adhere exactly to the target stem-loop structure required for toehold switch devices to function (Supplementary Fig. 8). Previous generative approaches to toehold switch design have approached this problem by manually correcting generated constructs that have been optimized using activation maximization, which introduces non-trivial changes to the predicted score36. Here, toehold switch generation was accomplished by adapting a thematically similar approach to previous RNA inverse design software, where a target secondary structure can be specified by a user. This took the form of a reverse complementation layer in the generator network which operated on the latent nucleotide array, mapping the regions upstream and downstream of the RBS that base pair in the toehold switch before being passed through a final convolutional output layer. This development allowed several improvements offered through differentiable design while converging efficiently (Supplementary Fig. 9) and retaining the ability to design molecules with a programmable target secondary structure.
We analyzed the resulting secondary structures of GARDN-generated toehold switches as well as those designed using activation maximization, constrained activation maximization (where the conserved sequence regions are included in the input seed)49,52, and NUPACK14. For a qualitative assessment of design quality, we computed the MFE structures of the toehold switch designs and overlaid them on top of one another with a degree of transparency to visualize the ensemble of secondary structures generated by the different design strategies (Fig. 5a–f). In this representation, the most likely sequential character at each position was additionally calculated, and the resulting overall structure dictated by this calculation is displayed in bold (a linear, unpaired structure is displayed for sequence groups that return an invalid structure). For quantitative assessment, we employed a metric that could provide single-nucleotide resolution of structural agreement based on the expected paired or unpaired state of each nucleotide across an entire group of sequences (see Methods). This structural agreement metric was used to score how well the toehold switches designed using each algorithm adhered specifically to the target canonical toehold switch structure (Fig. 5g, h). Sequences designed using NUPACK, without any added constraints on the target RNA sequence, showed the optimal structural distribution, as expected (Fig. 5a, h). The experimental toehold switch dataset used during the training process, which featured target RNA sequences from viruses and human genome sites, displayed lower structural agreement due to the additional sequence constraints but adopted the expected toehold switch secondary structure (Fig. 5b, h). In contrast, toehold switches designed using either standard or constrained activation maximization did not converge to a structure that would be expected to exhibit the key switching mechanism required to function (Fig. 5c, d), resulting in a significant reduction in structural agreement (Fig. 5h). Designs generated using activation maximization also failed to recapitulate the conserved RBS and start codon sequences necessary for translation (Supplementary Fig. 10). GARDN-generated toehold switches, however, demonstrated a steady increase in structural agreement over adversarial training iterations, until finally converging to a value which replicates the distribution of secondary structures of the experimental toehold switch dataset used during the training process (Fig. 5g, h). We also used GARDN in tandem with the SANDSTORM-toehold predictor to generate toehold switches optimized to have extreme ON values. These GARDN-SANDSTORM designs not only retained the ensemble secondary structures of the training dataset during optimization, but additionally incorporated specific sequence preferences that matched those previously correlated with high ON-state expression in experiments based on thermodynamic considerations (Fig. 5f, h and Supplementary Fig. 10). These results demonstrate that a combination of the adversarial training setup and optimization guided by a structurally informed predictive model can allow for thermodynamically realistic, high-performance sequences to be designed using GARDN models.
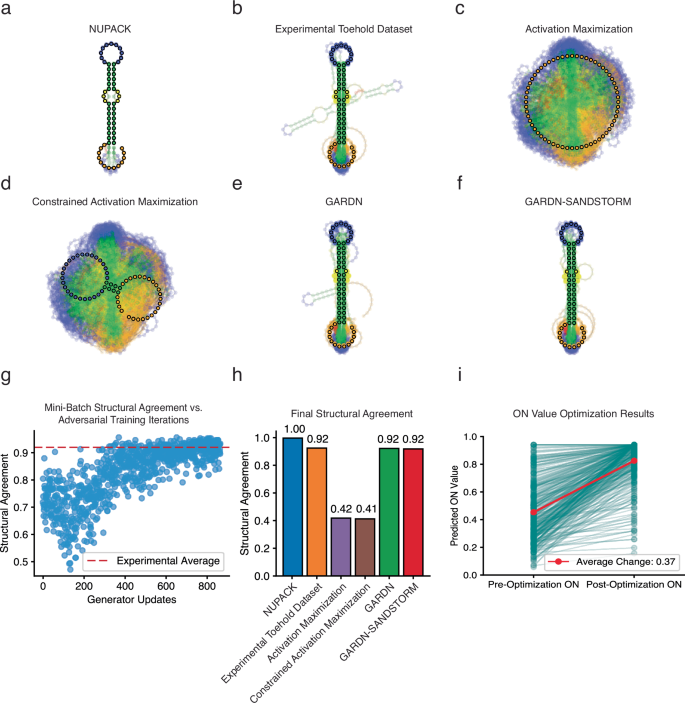
a–f Ensemble overlays of the secondary structures of toehold switch sequences designed using a variety of algorithms and those included in the experimental training data: (a) NUPACK design algorithm applied without target RNA sequence constraints, (b) the high-throughput experimental dataset35 used for model training, (c) single sequence activation maximization, (d) single sequence activation maximization with starting seeds containing the constant motifs, (e) GARDN, and (f) GARDN-SANDSTORM with toehold switches optimized to have high ON values (n = 300 a–f). Predicted structures were calculated using standard structural prediction software13,14,15, with the bolded structure representing the most likely structural character at each position (unpaired for heterogenous sequence groups where the most likely structure is not valid). g Structural agreement of GARDN-generated toehold switch sequences increases over adversarial training iterations, until converging to the experimental average. h Final structural agreement between toehold switches designed using different algorithms and the target canonical toehold switch secondary structure (quantification of a–f see Methods). i ON-value optimization results for GARDN-designed toehold switch sequences against the SANDSTORM-toehold predictive model. 300 calls to the predictive model resulted in a 37% increase in average ON score. Source data are provided as a Source Data file.
Previous approaches have relied on post-hoc editing of designed sequences to achieve a plausible binding structure. Introducing manual corrections after completing the optimization procedure introduces a non-trivial disconnect between the model’s prediction of the sequence’s function before and after the adjustment. By optimizing a generator with an output that adheres to the expected grammar of a toehold switch, the latent space providing inputs to the GARDN models can be efficiently explored with respect to ON values without having to make manual edits to the resulting sequence that are destructive to the validity of the prediction (Fig. 5f). The structural agreement traces of both GARDN and GARDN-SANDSTORM constructs (Supplementary Fig. 11) reveal that these sequences deviate from the target toehold switch structure at the same loci as the experimental dataset, demonstrating that both global and local secondary structural profiles are faithfully recreated by the generator. While sequences designed using NUPACK showed the optimal structural distribution, this effect is expected as the GARDN models were trained to recreate the experimental distribution (Fig. 5b) encountered during training.
We found that a pre-trained GARDN model also demonstrates better nucleotide variability (Supplementary Fig. 10) and faster generation time (Supplementary Fig. 11) than standard inverse design algorithms13,14,15, with the added benefit of having a score predicting design function. This variability is critical in in diagnostic applications where a limited set of solutions can impede the range of possible target molecules which activate the system and synthetic biology applications where high overlap between multiple constructs can lead to unintended cross-talk3. The lightweight SANDSTORM prediction architecture results in a simplified optimization landscape allowing for efficient and reliable sequence design. Using GARDN-SANDSTORM, we were able to achieve an average 37% percent increase in predicted normalized ON values of GARDN-generated sequences with only 300 calls to the predictive model (Fig. 5i). We repeated this procedure using several variations of the SANDSTORM architecture that varied in total parameter count, finding that models with a larger number of trainable parameters tended to over-estimate the level of improvement demonstrated between pre- and post-optimization sequences (Supplementary Fig. 12). Improving the predicted scores of generated constructs was possible without sacrificing the realistic sequence contents of the resulting designs (Supplementary Fig. 10). Toehold switches optimized by GARDN-SANDSTORM demonstrated superior variation in the trigger and stem domains compared to other approaches while maintaining the constant RBS and start codon motifs. Moreover, comparing the sequence contents of GARDN-optimized toehold switches to those optimized with standard activation maximization and constrained activation maximization demonstrates that in the absence of a generator network, optimized sequences will trend towards solutions that trivially maximize the score of the predictor at the expense of both sequence and structural design constraints (Fig. 5c, d).
Experimental validation of GARDN-SANDSTORM toehold switches
To verify the performance improvement afforded by GARDN-SANDSTORM generative RNA design, we experimentally characterized the performance of several groups of toehold switch sequences designed using different algorithms. These groups consisted of non-optimized outputs from the GARDN models, GARDN sequences optimized to have high ON-state GFP expression using an ON-only SANDSTORM predictive model, GARDN sequences designed to have both a high ON state GFP expression and low OFF state GFP expression using a joint ON/OFF SANDSTORM model, and those designed using NUPACK (see Source Data for sequences). For the most straightforward comparison between the design algorithms, we utilized the core NUPACK algorithm to design the 60-nt switch sequence only, rather than incorporating potential additional context such as the coding sequence or RBS, as the effect of incorporating these factors is not represented in the high-throughput training data. The resulting designs were characterized in Escherichia coli in conditions matching the toehold switch dataset used for training35 with the switch RNA on its own used to evaluate the OFF state and an RNA featuring a switch-trigger fusion used for the ON state (see Methods). Compared to the default GARDN output, sequences optimized with an ON-only SANDSTORM predictive model show a 3.8-fold increase in the geometric mean of GFP fluorescence from their parent sequences in the presence of the cognate trigger after only 300 calls to the predictive model (Fig. 6a; total optimization runtime ~11 s). The ON-optimized GARDN-SANDSTORM sequences additionally demonstrated an average fluorescence 4.8-fold higher than NUPACK (Fig. 6b). This result highlights the ability of our optimization process to improve specific attributes of RNA sequences.

a Individual toehold switch sequences designed by GARDN to have a high ON-state expression (see Supplementary Data 3 for sequences) show a geometric mean of 3.8-fold increase in GFP expression in the presence of their cognate trigger in E. coli after optimization by a SANDSTORM predictive model (p = 0.016, one-sided Wilcoxon ranked-sum test). b GARDN and ON-optimized GARDN-SANDSTORM toehold switches demonstrate a 1.9-fold and 4.8-fold increase in ON state fluorescence (p = 0.008, p = 0.002, one-sided Mann–Whitney U test). c, Toehold switches designed to exhibit high ON/OFF ratios by the GARDN-SANDSTORM routine demonstrate a geometric mean increase in ON/OFF ratio of 3.7-fold using 300 calls to a predictive SANDSTORM model (p = 0.016, Wilcoxon ranked-sum test). d Both non-optimized GARDN sequences and GARDN-SANDSTORM optimized groups show a significant increase in performance of 3.8-fold (p = 0.002, one-sided Mann–Whitney U test) and 11.9-fold (p = 0.002, one-sided Mann–Whitney U test), respectively, compared to those designed using classic inverse design software. See Methods (Flow Cytometry Analysis) and Supplemental Information for representative flow cytometry gating strategy. Bars represent the mean ± s.d. Source data are provided as a Source Data file.
Optimization of ON/OFF ratios is a more complex task, as the predictor must convey meaningful information related to the performance of each sequence in two independent experimental states. Furthermore, the sequence or structural factors that minimize leakage in the OFF state, such as high GC content that creates a strong stem, are frequently at odds to those that maximize expression in the ON state, where an AU-rich stem can more readily unwind leading to increased expression3. Despite this dynamic tradeoff, GARDN-SANDSTORM optimization by a predictor trained on the ON/OFF ratios of toehold switch sequences demonstrated a geometric mean of 3.7-fold increase in ON/OFF ratios compared to their non-optimized counterparts (Fig. 6c). These findings demonstrate the capability of the GARDN-SANDSTORM approach to optimize RNA molecules with complex, multi-faceted functions, a key factor for the design of RNA across modern biotechnology settings.
Both the non-optimized GARDN and GARDN-SANDSTORM sequence groups demonstrated remarkable improvements of 3.8-fold and 11.9-fold, respectively, in experimental ON/OFF ratios compared to those designed using NUPACK (Fig. 6d). Importantly, this increase in performance does not come at the expense of either the sequence or structural realism of the designs.
Screening a limited library of aptaswitches for SANDSTORM training
The use of generative modeling for molecular design is fundamentally constrained by the need to train on large quantities of experimentally collected data. This limitation can be prohibitive for the creation of new classes of riboregulators, in which only a handful of examples may show the desired regulatory effect in initial experiments. Despite this, there are many common design motifs that appear across a diverse range of functional RNA classes. For example, many recent works have utilized programmable RNA-RNA interactions to control cellular translation levels, or they have coupled toehold-switch-like secondary structures to the formation of a various regulatory motifs56,57,58,59,60,61. These systems have been engineered in settings ranging from cell-free systems to in vivo animal models. However, these same design motifs appear to consistently regulate diverse RNA processes. The use of generative modelling for these applications has thus far been limited by a lack of available training data.
We investigated if the design approach of coupling toehold switch-like structures to new regulatory motifs could be achieved by optimizing our GARDN model with a predictor trained in a new domain. To emulate settings in which high-throughput data may not be available, we used a low-throughput, manually collected library of 384 sequences. As the template for this investigation, we used the recently developed aptaswitch system58, which couples a stem-loop RNA secondary structure to the formation of a reporter aptamer via toehold-mediated strand displacement (Fig. 7a). Toehold switches and aptaswitches have common design features and critical differences that made them a plausible, yet challenging, candidate for our design strategy. Similar to the toehold switch, the aptaswitch stem-loop structure is unwound in response to a complementary RNA or DNA target. Unlike the toehold switch, however, the region of the aptaswitch released by target binding is used to form a downstream stem domain, promoting folding of the reporter aptamer and enabling binding of the ligand. For diagnostic purposes, the aptaswitch reporter aptamer is often Broccoli, which associates with its ligand DFHBI-1T to generate a bright green fluorescent signal62 Thus, output from the aptaswitches is not tied to translation of a reporter and aptaswitch activation requires a more complicated shift in structural conformations between the ON and OFF states compared to toehold switches. We constructed and screened an aptaswitch library by sampling 384 sequences from the Angenet-Mari et al.35 toehold switch dataset and converting these template hairpins into 137-nt aptaswitch constructs using the Broccoli aptamer as the reporter (Fig. 7b, c).

a Aptaswitches couple a toehold switch-like RNA hairpin to the conditional formation of a reporter aptamer. Fluorescence is generated in the presence of both the complementary target DNA molecule and the aptamer ligand. b A library of 384 aptaswitches were screened experimentally to evaluate the SANDSTORM model’s capacity on limited datasets. c The aptaswitch library was constructed by converting toehold switch sequences from Angenet-Mari et al35. into aptaswitches and manually characterized for their ON/OFF ratios (see Supplementary Data 4 for sequences). d The performance of converted aptaswitch elements varied greatly from their original context, demonstrating the need for a domain-specific predictor to be trained on the novel dataset. Toehold switch ON/OFF ratios are reported in the normalized value range of −1 to 1 from the original high throughput dataset. e The corresponding aptaswitch for all possible tiles of the covid N gene (n = 1230) were ranked in silico using SANDSTORM predicted ON/OFF ratios. The same process was repeated, instead using the NUPACK ensemble defect of the switch complex or switch-trigger complex to identify optimal regions from the same pool of candidate tiles. f The ON/OFF ratios of the top 6 aptaswitches identified using each approach are reported in addition to the 6 predicted lowest sequences identified using SANDSTORM. Bars represent the population mean ± s.d. Source data are provided as a Source Data file. Created in BioRender. Riley, A. (2025) https://BioRender.com/j80a514.
The converted aptaswitch library demonstrated a range of ON/OFF ratios spanning three orders of magnitude (Fig. 7b). While these devices contain many of the same structural elements as toehold switches, they were screened against free DNA targets and controlled a distinct biological output, resulting in performance that varied greatly from their original context (Fig. 7d). In order to apply our generative approach given this variation, a SANDSTORM predictive model needed to be characterized on the sequences as measured in their aptaswitch conformation. In data-scarce conditions such as these, deep learning predictive models are extremely susceptible to overfitting. While transfer learning can be efficiently used to fine-tune a pretrained predictive model on scarce examples in a new domain, we sought to investigate if the computational efficiency of the SANDSTORM architecture would enable us to train from scratch using this dataset. After benchmarking the model on 100 unique training-testing splits, we confirmed that the SANDSTORM-aptaswitch model reliably converged in 37 out of 100 training instances (Supplementary Figs. 13 and 14), achieving a median spearman rank correlation of 0.195 (Supplementary Fig. 13a, Appendix 3). We further validated that variation in predictive model performance was not due to favorable splitting of the data (Supplementary Fig. 13a–e, Appendix 3). This allowed us to confidently filter for models that demonstrated accurate training with minimal risk of selecting based on trivial performance driven by specific, favorable data samples.
We experimentally validated the predictive capacity of the trained SANDSTORM-aptaswitch models on unseen sequences in a setting emulating novel diagnostic design, where a high-performance aptaswitch sequence providing a high ON/OFF ratio against a specific pathogen target must be rapidly identified from a pool of hundreds of candidate sequences (Fig. 7e). A library of unseen aptaswitch sequences designed to recognize all 30-nt tiles of the SARS-CoV-2 N gene (N = 1230) was created in silico, and these sequences were ranked according to their predicted ON/OFF values using mean predictions across each of the successfully trained models. For comparison, we used NUPACK to create a ranked list of sequences from the same pool of possible tiles, using the ensemble defect of the switch (NUPACK Switch) or switch/trigger complex (NUPACK Complex) to rank preferrable sequences. The six highest predicted sequences for SANDSTORM demonstrated a mean ON/OFF ratio of 62.46, nearly tenfold higher than the mean ON/OFF ratio of the six predicted lowest of 6.69 (Fig. 7f, p = 0.0076, one-sided Mann–Whitney U test), confirming the performance of the model on unseen sequences despite the extreme scarcity of available data. In comparison, the top six sequences identified by NUPACK Switch and Complex approaches demonstrated mean ON/OFF ratios of 38.59 and 56.25 (Fig. 7f). SANDSTORM additionally identified the highest individual aptaswitch from the pool of tiles across each of the algorithmic approaches tested, a critical result for the deployment of this technology for rapid diagnostic design. Evaluating the entire N gene using a single SANDSTORM model required less than a second, while structural evaluations of this pool using NUPACK required 34.7 s. This efficiency is particularly relevant for real pandemic response settings, where the full-length genomes of multiple variants and closely related pathogens must be considered for diagnostic targeting on rapid timelines. This overall performance of SANDSTORM compared to thermodynamic tools demonstrates the potential for utilizing this framework to rapidly design RNA devices with limited experimental screening.
GARDN design of aptaswitches
To investigate whether the GARDN-toehold model could be optimized by the aptaswitch predictors to return high-performance sequences in this new domain, we conducted a computational sequence design experiment using each of the 37 successfully trained models. During optimization, the 60-nt toehold switch returned by the GARDN model was concatenated to the aptamer core and stem-forming b domain before being scored by a specific SANDSTORM predictor (Fig. 8a). Each SANDSTORM model was tasked with independently optimizing 100 converted aptaswitch sequences returned by the GARDN-toehold model for 300 optimization steps per sequence. From this group, we identified the specific model that demonstrated the greatest improvement between the pre- and post-optimized sequences (Model A) as ranked by the mean predicted ON/OFF of the 36 other models to carry forward for experimental validation. We hypothesized that the consensus between models regarding the performance of sequences designed under the guidance of model A would translate to the largest improvement in experimental performance. The GARDN-SANDSTORM optimized sequences demonstrated a geometric mean improvement in ON/OFF ratio of 1.49 across twelve pre- and post-optimization aptaswitches (Fig. 8b), with the post-optimization group demonstrating a mean ON/OFF ratio of 65.21 compared to the pre-optimization average of 40.45 (p = 0.1, one-sided Wilcoxon signed-rank test). For comparison, we utilized NUPACK to design a group of 12 aptaswitches without any additional sequence constraints, which demonstrated a similar mean ON/OFF ratio of 67.92 (Fig. 8b).

a The pretrained GARDN-toehold model was optimized by a SANDSTORM predictor trained on aptaswitch performance. The GARDN model generates 60-nt toehold switch hairpins from a random variable (Z), which were manually concatenated to the aptamer core as well as the necessary repeat of the (b) domain for aptamer formation during runtime. b The ON/OFF ratios of sequences designed using NUPACK, GARDN-SANDSTORM, and GARDN-SANDSTORM under an extended optimization routine (see Supplementary Data 5 for sequences). Data points are the normalized ON/OFF ratios for individual aptaswitch sequences. Bars are the population mean ± s.d. c The ON/OFF ratios of the paired pre- and post-optimization samples designed by SANDSTORM model A for 300 optimization steps. 7/12 samples show an improved post-optimization ON/OFF value. d The ON/OFF ratios for paired samples resulting from 600 optimization steps under the guidance of model A. 10/12 samples show an improved post-optimization ON/OFF value. Bars indicate the normalized ON/OFF value extrapolated from the ON and OFF state measurements demonstrated at three different aptaswitch concentrations. Source data are provided as a Source Data file.
Unlike in our investigation of toehold switches and RBSs, where every optimized sequence showed improvement, we found that 7/12 sequence optimization pairs demonstrated improvements for model A under identical optimization conditions to those explored throughout our previous experiments (Fig. 8c). While this change was not as drastic as the case of using SANDSTORM alone to screen aptaswitch candidates, this was to be expected given that the pre-optimization sequences were not initially selected for low performance, but were random. We further explored if a prolonged optimization relative to the initial settings could improve the downstream performance of the returned aptaswitches. We increased the number of steps in the GARDN-SANDSTORM design process from 300 to 600, again using model A to guide the procedure. Under these conditions, the post-optimization group demonstrated a mean ON/OFF ratio of 74.02 compared to the average of 52.57 for the initial group (p = 0.039, one-sided Wilcoxon signed-rank test) with 10/12 sequences improving. The geometric mean of the improvement demonstrated by optimized sequences was 2.07, (Fig. 8d) indicating the utility of this approach despite the limited availability of data. The highest ON/OFF ratios for the post-optimized GARDN-SANDSTORM aptaswitch groups were 161.9× and 158.3×, despite encountering only 14 sequences (3.6% of the dataset) greater than or equal to this activity during training. These results demonstrate the potential for SANDSTORM and GARDN models to serve as valuable components of the RNA design toolkit regardless of the functional complexity or data limitations of the design task.
Discussion
We have demonstrated that by incorporating the secondary structure of RNA molecules into a deep learning framework, we can achieve state-of-the-art functional prediction performance using a streamlined, generalizable CNN architecture. Additionally, we have shown that these models can extract interpretable abstractions of RNA structure—the presented generative models built around this encoding recreate experimentally validated thermodynamic properties while being optimized for function. These properties directly translate to improved experimental performance compared to current state-of-the-art sequence design tools while retaining critical conserved sequence elements, nucleotide diversity, and targeted structural properties.
The GARDN approach offers a strong solution for designing structurally realistic RNA molecules with functions that cannot be accurately predicted using thermodynamic attributes. It is particularly helpful in domains that offer limited training data, or where candidate molecules must adopt a target secondary structure in a sequence-independent manner. Future investigations could explore if this design paradigm shows improved performance by avoiding the underlying thermodynamic assumptions of standard inverse design tools or by accounting for additional factors that may be represented in high-throughput data. GARDN-SANDSTORM optimization pairs additionally outperform the 2.6-fold improvement in ON/OFF ratios reported for activation maximization sequences from previous studies36 while limiting optimization runtime, which could be the product of the elimination of post-hoc sequence adjustments or improved optimization efficiency given the streamlined predictors.
Both SANDSTORM and GARDN models could be readily applied to new datasets mapping RNA variants to their functional performance. Other domains such as transcriptional riboregulators, IRES-like elements, and RNA protein-binding recognition sites63,64,65,66 all have a critical dependency on RNA structure that would make them aptly suited for SANDSTORM predictions and GARDN design. While there is no definitive lower bound on the number of sequences needed to deploy these models, the results demonstrated on our aptaswitch dataset suggest that these tools may be extremely useful for designing new RNA devices in data-scarce conditions. This capability could prove valuable in future pandemic response contexts, where rapidly identifying an optimal diagnostic with limited available data is common, or as a copilot for designing novel riboregulators containing toehold switch design motifs. The efficiency of the SANDSTORM models enabled meaningful identification of sequence-to-function relationships that could be leveraged for design using only 384 training examples. Future areas of investigation in data-scarce conditions could explore unsupervised identification of adequately trained models that are propagated for downstream design tasks or could explore the possibility of expanding upon the strict optimization limit of only 600 predictor queries imposed on our framework during the design and optimization procedure.
We expect that future work will focus on striking the same balance between biological-feature engineering and deep-learning-based feature abstraction that enable the enhanced performance of the SANDSTORM models. While the predictive capability demonstrated here could likely be improved by fine-tuning the dual-input architecture to each task, retaining generalizability is a critical constraint for the design of machine learning systems in biological contexts. The expansion of the latent reverse complementation layer of the GARDN-toehold generator to an arbitrary target secondary structure could additionally enable the benefits of differentiable design for several de novo RNA devices. In this work we have focused on relatively short functional RNA sequences up to 137 nt in length and on datasets of uniform RNA length. As the toolkit of RNA molecules expands in both length and complexity and more RNA libraries of varying sequence length are tested, further investigation will be needed to overcome the challenges these tasks will inevitably present.
As engineered functional RNAs continue to gain traction in various domains of biotechnology, the toolkit of computational methods for RNA design must keep pace. Reliable generative tools for engineering functional sequences will continue to be essential for rapidly adapting RNA systems to new use cases with minimal experimental screening. SANDSTORM and GARDN models offer a promising solution to the experimental screening challenge; the generalized ability to predict and design functional RNA molecules with increased performance while maintaining realistic secondary structures could serve as the foundation of a next-generation set of computational RNA tools.
Methods
Secondary structure array calculation
The secondary structure array presented in this work is an \({NxN}\) array, where \(N\) is the length of an RNA sequence. Each point \((i,j)\) in this array is assigned a value based on the possibility of the nucleotide at position \(i\) forming a Watson-Crick base pair with the nucleotide at position \(j\). If \((i,j)\) does not form a canonical base pair, a value of 0 is assigned. If \((i,j)\) forms an A-U base pair or G-U wobble base pair, a value of 2 is assigned, and if \((i,j)\) forms a G-C base pair, a value of 3 is assigned. These values reflect the number of hydrogen bonds that form in their respective base pairing interactions and are included to provide the model with information related to binding strength.
Structure-based classification testing
Several classes of labelled decoy toehold switches were manually constructed by inserting conserved sequence elements at the required positions and creating the necessary one-hot and structural arrays. These sequences were used to evaluate the ability of the structural array to encode meaningful information. In the first training experiment (Fig. 2), the dataset consisted of 8000 unique, one-hot-encoded sequences for each decoy class combined with 8000 original toehold switches sampled randomly from the Angenent-Mari et al.35 dataset. Standard multi-class classification was conducted on this synthetic dataset using a simplified version of both the two-input and sequence-only models and multi-categorical cross-entropy loss. The introduction of a second convolutional channel in the SANDSTORM model by default will increase the number of trainable parameters relative to a sequence-only model, which may lead to an artificial inflation in the performance of the two-channel models. To compensate for this, the two-input and sequence-only models were composed of the same depth of convolutional layers (3), but the number of filters per convolutional layer in the dual-input model was reduced to balance the number of trainable parameters between the two. The same split of data (75% training, 25% testing) was used to compare the performance of the two models, with ROC curves and AUC metrics calculated using sklearn. To investigate the impact of receptive field size on sequence or paired input model performance, the same predictive models and training data as before were employed, however, the first convolutional filter varied in size beginning with a 1 × 1 kernel and then exploring kernels ranging from 4 × 1 to 4 × 30 in increments of 1. To explore the performance of the models under varying total parameter counts, the number of filters per convolutional layer in each layer of the models was increased while maintaining an initial filter size of 4 × 18. The data ablation investigation was conducted by increasing the number of RBS-AUG decoys in the dataset from 2000 to 48000, with the AUC values for this class specifically reported. Across each of these accessory tests, the same train-test splits were supplied to both the sequence and paired input models in three independent, randomized replicates.
SANDSTORM predictive architecture and training
The full-sized predictive model consisted of two parallel stacks of three convolutional layers, with each convolutional stack extracting motifs from the one-hot-encoded sequence arrays and structural arrays, respectively. The structural array convolutional channel consisted of a spatial dropout layer (dropout = 0.2) between the first and second convolutional operations while the sequential input channel included batch normalization at this layer. Global maximum pooling was applied to the output of the structural convolutional stack (GlobalMaxPooling2D within tensorflow). The outputs of these convolutions were concatenated before being passed through a series of three densely connected layers to a final output that returned a value for each functional property of the input sequences (e.g., ON and OFF for toehold switches, Mean Ribosome Loading for UTRs, etc.). Relu internal activation functions were used for all layers of the model and training was conducted using the Adam optimizer.
The model was retrained for each published dataset without hyperparameter tuning to ensure generalizability across classes. While hyperparameter tuning was not conducted, the resulting model size for each task does vary slightly due to the variation in size of the input sequence. Model training was conducted in the same environment for both the SANSTORM and previously published model in each case (see Supplementary Fig. 1 for a detailed diagram of the model architecture). For toehold switches, RBSs, and 5′ UTR training, both the SANDSTORM model and comparator were trained on three randomly identified training-testing splits using the sklearn toolkit (train_test_split, test size = 20%) while stratifying with respect to the target variable. For gRNA data splitting, the code from the original work was utilized to identify training splits as inputs to the previously published model based on the resampled dataset with the following parameters: subset=’exp’, stratify_by_pos=False. Data splits for the SANDSTORM gRNA predictor were identified using a grouped splitting (sklearn, GroupedShuffleSplit) function to ensure that complete groups of resampled elements were retained uniquely within either the training or testing sets. Reported metrics were averaged across each of the training replicates. The SANDSTORM gRNA predictor was additionally expanded to accept the paired guide-target encoding employed by the original work38. In this context, the structural array inputs were calculated based on the sequence of the target RNAs with 20 nt of additional context. Jupyter Notebooks allowing the retraining of both SANDSTORM and the comparator model from scratch within the same environment are available in the code repository. A refined training procedure was conducted on the limited aptaswitch dataset that is outlined in detail in Appendix 3.
GARDN architecture
The GARDN-toehold generator models were trained under WGAN-GP settings. The GARDN-toehold generator was composed of a series of upsampling layers followed by spectrally normalized convolutional layers and batch normalization. This layer progression was stacked 3 times, with upsampling layer sizes of (2,5), (2,6), and (1,2), eventually returning a 60-nt sequence. As the original toehold switch dataset consisted of 59 variable nucleotides, real samples were concatenated to the 5′ conserved ‘C’ nucleotide of the pre-sequence linker to allow the length to be a non-prime number for the upsampling progression. Following these upsampling operations was the programmable reverse complementation layer broadcasting nucleotide positions and a final convolutional layer. The toehold-discriminator model consisted of 3 spectrally normalized convolutional layers, each followed by a layer normalization operation as a drop-in replacement for batch normalization as suggested by the original WGAN-GP authors55. The GARDN-UTR model leveraged transpose convolutional layers in place of upsampling with sizes of (2,5), (2,5), and (1,2) to reach the 50-nt final sequence output. The GARDN-UTR generator also utilized a self-attention layer67 following the three transposed convolutions and before being passed through the final convolutional output instead of a programmable reverse complementation operation to potentially allow for non-specific, long-range nucleotide dependencies. The GARDN-RBS model followed a similar architecture that included a self-attention layer; however, the final sequences were 17 nt in length. A layer progression outputting an 18 nt sequence array was deployed ((2,3), (2,3), (1,2)), with a slicing operation utilized to extract all but the first nucleotide, which was not considered by the discriminator. This resulted in the necessary 17 nt array that could be accepted as an input by the SANDSTORM-RBS predictor. The full model details, trained generative components, and scripts for retraining can be found in the code repository.
GARDN reverse complementation layer
A reverse complementation layer was developed to allow GARDN models to generate sequences adhering to the target structural grammar defining toehold switches. In this approach the one-hot-encoded values returned by the final upsampling layer in the 5′ portion of the stem region of the toehold switch were reversed along their second (nucleotide) and third (sequence position) axes before being broadcast to the necessary 3′ portion of the stem region and passed through a final convolutional layer. This method greatly improved the model’s ability to generate sequences with canonical toehold switch structures and in the future could be generalized to model arbitrary target RNA structures while maintaining valuable nucleotide diversity.
GARDN optimization
In a standard activation maximization setup, the input, \(X\), to a differentiable predictive model, \({P}_{\delta }\), is repeatedly summed with its own gradients to maximize the returned value while the parameters of the predictive model, \(\delta\), are held constant. This can be formalized as
$$X\to X+{{{{\rm{\alpha }}}}\nabla }_{X}{P}_{\delta }(X),$$
(1)
where \({{{\rm{\alpha }}}}\) is a constant that is analogous to the learning rate of a standard neural network. In the case of sequence design, the inputs are a one-hot-encoded nucleotide array passed through some differentiable argmax operation (SoftMax, Gumbel SoftMax, Relaxed One-Hot Encoding) to preserve the discretized inputs expected by the predictive model while still allowing continuous gradient steps to take effect. To optimize GARDN outputs, we adapted the method from Killoran et al.45, where instead of directly optimizing the nucleotide array input to the predictive model, we take the gradient of the random variable seed, \(Z\), of our generative model with respect to the predictive output, while freezing the weights of both the predictive and generative models (\(\delta\) and \(\theta\)). This approach allows us to move through the latent space providing input seeds to the generator until a region is found that returns sequences with desirable features as measured by the SANDSTORM predictor.
$$Z\to Z{+{{{\rm{\alpha }}}}\nabla }_{Z}{P}_{\delta }({G}_{\theta }\left(Z\right))$$
(2)
$$Z\to Z{-{{{\rm{\alpha }}}}\nabla }_{Z}{P}_{\delta }({G}_{\theta }\left(Z\right))$$
(3)
In this setup, the gradient step can either be summed with or subtracted from the original seed to move towards regions of the latent space that correspond to the generation of high- or low-performing sequences. At each gradient update step, the structural array input corresponding to the generated sequence is calculated and fed in parallel to the frozen SANDSTORM model; a key step that requires the memory and compute efficiencies of our novel structural array. For our study the predictor returned values related to the experimental function of RNA molecules; however, this predictor could be generalized to any arbitrary differentiable function.
Computational sequence design experiments
To design UTRs, we generated 300 high-expression UTR sequences using gradient ascent on latent inputs to the generator and 300 low-expression sequences by using gradient descent (subtraction) on latent inputs to the generator. We compared the nucleotide contents and thermodynamic profiles of these generated constructs to the highest performing (mean ribosome loading > 8.5) and lowest performing (mean ribosome loading <1.2) UTRs from the experimental dataset. For toehold switches, the predictive model returns two distinct outputs, one for ON and OFF. We chose to design 300 toehold switches with a maximized predicted ON value and compared to those designed by standard thermodynamic inverse design via NUPACK, single-sequence activation maximization, and single-sequence activation maximization with starting seeds that already contained the start codon and RBS motifs (constrained activation maximization). The GARDN-optimized sequences were designed using our novel two-input model, while both activation maximization approaches were designed by performing gradient ascent of a single sequence (n = 300 total sequences) using the predictive model from Valeri et al.36. The sequence logos demonstrated throughout this work represent the average nucleotide frequencies of these groups of sequences. For RBS sequences, 100 candidates were designed using the GARDN-SANDSTORM routine using 300 update calls to the predictive model, with those exhibiting the highest delta between pre- and post-optimized expression selected for testing.
Toehold switch library design
A library of six toehold switch sequences were selected from several design approaches to validate the performance improvements offered through GARDN-SANDSTORM. To replicate the conditions of the high-throughput dataset used to train the SANDSTORM models, ON-state sequences were validated in the switch-trigger fusion configuration originally used by Angenet-Mari et al.35. For the most direct performance comparison to the previously published work, the SANDSTORM predictor shown in Fig. 3 and the one used for the computational optimization in Fig. 5 matched the formatting of the original work, where a single model predicts both the ON and OFF values of each sequence. In practice however, we found a substantial increase in performance by training on the predicted ON/OFF ratio itself as a single output, which we utilized to optimize the experimentally validated sequences. The first group consisted of six sequences generated randomly from GARDN outputs. Additionally, 100 GARDN sequences were optimized by an ON-only predictive SANDSTORM model (300 optimization calls), with the six sequences that demonstrated the highest predicted increase in ON state selected for experimental validation. This approach was repeated to select an additional six sequences with a SANDSTORM model that predicted the ON/OFF ratios. A final set of six sequences was designed using the inverse-design toolkit offered by NUPACK 4.0.0.28 under default model conditions14. For this group, inverse design was carried out by individually specifying the switch complex without any sequence constraints beyond the RBS, start codon, and reporter gene sequences.
Plasmid construction
All toehold switch sequences were ordered as single-stranded DNA oligonucleotide ultramers from Integrated DNA Technologies with common 30-nt homology regions (Supplementary Data 6). PCR reactions were carried out with Q5 High-Fidelity 2× Master mix (New England Biolabs, M0492S) using universal primers (Supplementary Data 6) to create double-stranded DNA, and the amplified DNAs were connected with plasmid backbones using Gibson assembly (NEBuilder HiFi DNA Assembly Master Mix, E2621S). Plasmid backbones were amplified from the commercially available pCOLADuet (EMD Millipore) expression vector via PCR followed by digestion with the restriction enzyme DpnI (New England Biolabs, R0176S). All Gibson assembly products were transformed directly into E. coli BL21 (DE3) cells (New England Biolabs, C2527H), and sequence verified by Sanger sequencing.
GARDN-designed RBS and SANDSTORM-RBS-optimized sequences were integrated into the pCOLADuet vector by Q5 site-directed mutagenesis (New England Biolabs, E0554S). Briefly, forward and reverse PCR primers were designed to anneal to the linker region of the toehold switch and T7 promoter, respectively3. The forward primer bound to the linker (AACCTGGCGGCAGCGCAAAAG) and contained each 17-nt RBS design, followed by a conserved 6-nt spacer (CATAAC) and start codon (Supplementary Data 6). The universal reverse primer bound to the T7 promoter and featured a 36-nt spacer overhang. Following PCR, the amplicons were subject to a KLD reaction (New England Biolabs, M0554S), transformed into E. coli 5-apha (New England Biolabs (C2987H), and sequence verified by Sanger Sequencing. Plasmids were then transformed into BL21 (DE3) cells (New England Biolabs, C2527H). The reporter protein utilized in the study is GFPmut3b with an ASV degradation tag.
Flow cytometry analysis
Bacterial colonies transformed with plasmids containing either the fused switch-trigger construct (ON state) or with just the unfused switch (OFF state) were inoculated in 1 mL of LB (Sigma, L3522) in triplicate with 50 µg mL−1 kanamycin antibiotic (Sigma K1377) and grown overnight at 37 °C shaking at 750 rpm. After 14 h, 5 μL of overnight culture was diluted 100-fold in 495 μL of fresh LB with 25 µg mL−1 kanamycin. Following recovery until OD600 reached approximately 0.2, isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma I6758) was added to each well to a final concentration of 0.25 mM.
After 3 h of induction with IPTG, flow cytometry was performed with a Stratedigm S1000 cell analyzer equipped with an A600 Stratedigm high-throughput auto sampler. Prior to running flow cytometry, inoculated cultures were diluted twofold into phosphate buffered saline (Sigma, P4417) in 384-well plates. Approximately 50,000 individual cells were recorded per sample, and cell populations were gated according to their forward side scatter (FSC) and side scatter (SSC) distributions3,8. (see Supplementary Fig. 15 for representative gating data). The E. coli population exhibited a single peak in the two-dimensional histogram due to unimodal distributions in both FSC and SCC. To establish the gating area, the maximum histogram value within this peak was identified, and coordinates in the histogram where values were at least 15% of this maximum were selected using FlowJoTM v10.8 Software (BD Life Sciences). Following gating by FSC-A and SSC-A, the selected population was subject to gating among the FSC-A and FSC-H parameters to select for single cells. The resulting mean GFP fluorescence signal generated by these gated populations was used for subsequent calculations. Fluorescence measurements of ON-state and OFF-state cells were calculated as the mean of three technical replicates, and error levels were calculated from the standard deviation of measurements from three biological replicates (nine samples total per construct).
Aptaswitch library design
The library of aptaswitches was created by converting randomly sampled toehold switches from the Angenet-mari et al.35 dataset into aptaswitch structures. To ensure the full range of toehold switch performance was represented in our adapted library, the 20 highest and 20 lowest performing sequences in the original screen were selected in addition to 344 random samples. The first seven nucleotides of the sequestering hairpin were considered the b domain; to construct an aptaswitch the standard Broccoli core aptamer sequence was concatenated to the 3′ end of the toehold switch followed by a repeat of the b domain which forms the stabilizing hairpin in the ON conformation.
In vitro transcription and experimental screening of aptaswitches
All single-stranded DNA oligonucleotides were purchased from Integrated DNA Technologies. Aptaswitch RNAs were in vitro transcribed at 37 °C for 4 h using AmpliScribeTM T7-FlashTM Transcription Kits (Lucigen, ASF3507) with 400 nM of the DNA template. All transcription products were purified using Zymo Research RNA Clean & Concentrator-96 (Zymo Research R1080) and quantified using a BioTek Take3 Trio Microvolume Plate and a BioTek Cytation 5 Cell Imaging Multimode Reader.
Prior to measurement, three 1:2 serial dilutions of all aptaswtich devices were performed in RNase/DNase free water (Thermo Scientific 10977023). 40 μM DFHBI-1T (Lucerna, 410) was prepared in a buffer consisting of 40 mM HEPES Buffer pH 7.4 (Fisher Scientific, AAJ67502), 100 mM KCL (Sigma, 60142), and 5 mM MgCl2 (Sigma, M1028) filter sterilized with a syringe fixed with a 0.22 μM filter (Celltreat 229743) and stored in a light-protective 1.7 mL tube. Final 35 μL reactions were assembled as follows in a 384 well plate (Corning 3540): 2 μM DFHBI-1T buffer, 1.75 μL of aptaswtich device at each concentration, and 15 μM cognate target single stranded DNA. The volume was brought to 35 μL with RNase/DNase free water. A BioTek Synergy Neo2 multimode microplate reader was used for measurement. Before each measurement, the instrument was warmed to 37 °C and samples were shaken linearly for 30 s to ensure proper mixing. Measurements were taken for 4 h at excitation 472/15, emission 507/15 from the bottom of the plate with a gain of 85.
Aptaswitch data preparation
Each aptaswitch was measured individually in the OFF state, where the cognate trigger was absent but DFHBI-1T was present, and the ON state, where both the trigger and DFHBI-1T were present. Fluorescence values were calculated as the mean intensity measured 3.5 h after the start of the assay, using a window of 50 discrete sampling time points to compute. To account for the variation in aptaswitch RNA concentration following in vitro transcription, which ranged in magnitude from <100 nm to 1000 nm, we computed a normalized ON/OFF value for each switch by measuring independently the ON and OFF values at three different concentrations, and extrapolating a normalized value for each state from the linear fit of these curves. The three concentration values utilized for fluorescence quantification were (1) the non-diluted IVT product, (2) a 2× dilution of the IVT product and (3) a 4× dilution of the IVT product. In the ON state, fluorescence increased linearly with concentration (Supplementary Fig. 16), while in the OFF state, a subset of switches showed near background fluorescence levels across concentration values. For those switches which demonstrated a linear increase in fluorescence as concentration increased in the OFF state, the same linear fitting approach was applied to identify a normalized OFF value. For those which demonstrated near-background levels across concentrations, the mean OFF value across the three measurements was utilized. This approach creates a subset of aptaswitches that will show a linear increase in ON/OFF values with increasing concentration due to the constant OFF state; to mitigate the possibility of inflationary effects of this normalization we utilized a value of 500 nM to calculate normalized concentrations. The log-transformed ON/OFF value was calculated and passed through sklearn standard scaling before being supplied to the model for training.
Thermodynamic parameter calculations
All results demonstrating the thermodynamic properties of sequences were calculated using NUPACK 4.0.0 in python under default model settings. We computed a sliding window minimum free energy calculation (Supplementary Fig. 7) by slicing out varying sizes of the sequences to plot traces of localized secondary structures and GC content. Distributions of ensemble defect are calculated across the entire sequence.
Structural agreement
In the case of toehold switches, a more specific quantification of secondary structure was needed to analyze the performance of the GARDN models than in the case of UTRs or RBSs. To do so we employed the ‘structural agreement’ metric that reflects how well a dataset of generated sequences adheres to a specific secondary structure. This value is obtained by computing the predicted MFE secondary structure of every sequence in a particular dataset in dot-bracket notation. In this notation, each position in a sequence can take the value of ‘.’ to represent an unpaired nucleotide, ‘(‘ to represent a nucleotide bound to a downstream nucleotide, and ‘)’ to represent a nucleotide bound to an upstream nucleotide. The frequencies of each of symbol at each position can be averaged across a dataset to form a consensus structural array representing the probability of finding each symbol at each position (Supplementary Fig. 17). The argmax can be computed along this array of structural probabilities to return the positional consensus secondary structure. For canonical toehold switches, the target secondary structure in dot-bracket notation is ‘…………(((((((((…((((((………..))))))…)))))))))’. We compute the structural agreement for a dataset of sequences by the following:
$$\frac{1}{N}{\sum }_{i=1}^{N}p(i),$$
where \(N\) is the length of the target secondary structure and \(p(i)\) is the probability of the base at position i adopting the dot-bracket symbol that matches that of the target secondary structure. The values for \(p(i)\) are calculated from the consensus structural array. Without averaging, \(p(i)\) can be plotted as a trace to provide single-nucleotide resolution of where a particular dataset diverges from the target structure (Supplementary Fig. 11).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. The computational datasets used for predictive model comparisons can be found at the original source references (summarized in Supplementary Data 1). Summarized experimental measurements from all model designs including sequence information can be found in Supplementary Data 2–5. Source data are provided with this paper.
Code availability
The code used to develop the model, perform the analyses and generate results in this study is publicly available and has been deposited in at https://github.com/AlexGreenLab/GARDN-SANDSTORM under an academic license68.
References
Damase, T. R. et al. The Limitless Future of RNA Therapeutics. Front Bioeng. Biotechnol. 9, 628137 (2021).
Pickar-Oliver, A. & Gersbach, C. A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507 (2019).
Green, A. A., Silver, P. A., Collins, J. J. & Yin, P. Toehold switches: de-novo-designed regulators of gene expression. Cell 159, 925–939 (2014).
Bayer, T. S. & Smolke, C. D. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nat. Biotechnol. 23, 337–343 (2005).
Ma, D. et al. Multi-arm RNA junctions encoding molecular logic unconstrained by input sequence for versatile cell-free diagnostics. Nat. Biomed. Eng. 6, 298–309 (2022).
Castillo-Hair, S. M. & Seelig, G. Machine learning for designing next-generationmRNA therapeutics. Acc. Chem. Res. 55, 24–34 (2022).
Kim, J. et al. De novo-designed translation-repressing riboregulators for multi-input cellular logic. Nat. Chem. Biol. 15, 1173–1182 (2019).
Green, A. A. et al. Complex cellular logic computation using ribocomputing devices. Nature 548, 117–121 (2017).
Chappell, J., Westbrook, A., Verosloff, M. & Lucks, J. B. Computational design of small transcription activating RNAs for versatile and dynamic gene regulation. Nat. Commun. 8, 1051 (2017).
Hu, C. Y., Varner, J. D. & Lucks, J. B. Generating effective models and parameters for RNA genetic circuits. ACS Synth. Biol. 4, 914–926 (2015).
Salis, H. M., Mirsky, E. A. & Voigt, C. A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 27, 946–950 (2009).
Orlandini von Niessen, A. G. et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3′ UTRs identified by cellular library screening. Mol. Ther. 27, 824–836 (2019).
Dirks, R. M., Lin, M., Winfree, E. & Pierce, N. A. Paradigms for computational nucleic acid design. Nucleic Acids Res. 32, 1392–1403 (2004).
Fornace, M. E., Porubsky, N. J. & Pierce, N. A. A Unified Dynamic Programming Framework for the Analysis of Interacting Nucleic Acid Strands: Enhanced Models, Scalability, and Speed. ACS Synth Biol 9, 2665–2678 (2020).
Zadeh, J. N., Wolfe, B. R. & Pierce, N. A. Nucleic acid sequence design via efficient ensemble defect optimization. J. Comput. Chem. 32, 439–452 (2011).
Churkin, A. et al. Design of RNAs: comparing programs for inverse RNA folding. Brief. Bioinforma. 19, 350–358 (2018).
Zhang, H., Li, S., Zhang, L., Mathews, D. H. & Huang, L. LazySampling and LinearSampling: fast stochastic sampling of RNA secondary structure with applications to SARS-CoV-2. Nucleic Acids Res. 51, e7 (2023).
Zhang, H. et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature 621, 396–403 (2023).
Singh, J., Hanson, J., Paliwal, K. & Zhou, Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat. Commun. 10, 5407 (2019).
Sato, K., Akiyama, M. & Sakakibara, Y. RNA secondary structure prediction using deep learning with thermodynamic integration. Nat. Commun. 12, 941 (2021).
Zhang, J., Fei, Y., Sun, L. & Zhang, Q. C. Advances and opportunities in RNA structure experimental determination and computational modeling. Nat. Methods 19, 1193–1207 (2022).
Sumi, S., Hamada, M. & Saito, H. Deep generative design of RNA family sequences. Nat. Methods 21, 435–443 (2024).
Schmidt, C. M. & Smolke, C. D. A convolutional neural network for the prediction and forward design of ribozyme-based gene-control elements. eLife 10, e59697 (2021).
Linder, J., Koplik, S. E., Kundaje, A. & Seelig, G. Deciphering the impact of genetic variation on human polyadenylation using APARENT2. Genome Biol. 23, 232 (2022).
Townshend, B., Xiang, J. S., Manzanarez, G., Hayden, E. J. & Smolke, C. D. A multiplexed, automated evolution pipeline enables scalable discovery and characterization of biosensors. Nat. Commun. 12, 1437 (2021).
Leppek, K. et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat. Commun. 13, 1536 (2022).
Wayment-Steele, H. K. et al. Deep learning models for predicting RNA degradation via dual crowdsourcing. Nat. Mach. Intell. 4, 1174–1184 (2022).
Cao, J. et al. High-throughput 5′ UTR engineering for enhanced protein production in non-viral gene therapies. Nat. Commun. 12, 4138 (2021).
Townshend, B., Kennedy, A. B., Xiang, J. S. & Smolke, C. D. High-throughput cellular RNA device engineering. Nat. Methods 12, 989–994 (2015).
Wayment-Steele, H. K. et al. RNA secondary structure packages evaluated and improved by high-throughput experiments. Nat Methods 19, 1234–1242 (2022).
Ackerman, C. M. et al. Massively multiplexed nucleic acid detection with Cas13. Nature 582, 277–282 (2020).
Xiang, J. S. et al. Massively parallel RNA device engineering in mammalian cells with RNA-Seq. Nat. Commun. 10, 4327 (2019).
Lee, J. et al. RNA design rules from a massive open laboratory. Proc. Natl. Acad. Sci. USA 111, 2122–2127 (2014).
Wayment-Steele, H. K. et al. RNA secondary structure packages evaluated and improved by high-throughput experiments. Nat. Methods 19, 1234–1242 (2022).
Angenent-Mari, N. M., Garruss, A. S., Soenksen, L. R., Church, G. & Collins, J. J. A deep learning approach to programmable RNA switches. Nat. Commun. 11, 5057 (2020).
Valeri, J. A. et al. Sequence-to-function deep learning frameworks for engineered riboregulators. Nat. Commun. 11, 5058 (2020).
Sample, P. J. et al. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 37, 803–809 (2019).
Metsky, H. C. et al. Designing sensitive viral diagnostics with machine learning. Nat. Biotechnol. 40, 1123–1131 (2022).
Höllerer, S. et al. Large-scale DNA-based phenotypic recording and deep learning enable highly accurate sequence-function mapping. Nat. Commun. 11, 3551 (2020).
Aloysius, N. & Geetha, M. A review on deep convolutional neural networks. In 2017 International Conference on Communication and Signal Processing (ICCSP) 0588–0592 https://doi.org/10.1109/ICCSP.2017.8286426 (2017).
Sundararajan, M., Taly, A. & Yan, Q. Axiomatic attribution for deep networks. in Proceedings of the 34th International Conference on Machine Learning (eds. Precup, D. & Teh, Y. W.) - Volume 70 3319–3328 (JMLR.org, Sydney, NSW, Australia, 2017).
Mauger, D. M. et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 116, 24075–24083 (2019).
Gingold, H. & Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 7, 481 (2011).
Espah Borujeni, A., Channarasappa, A. S. & Salis, H. M. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 42, 2646–2659 (2014).
Killoran, N., Lee, L. J., Delong, A., Duvenaud, D. & Frey, B. J. Generating and designing DNA with deep generative models. Preprint at https://doi.org/10.48550/arXiv.1712.06148 (2017).
Castro, E. et al. Transformer-based protein generation with regularized latent space optimization. Nat. Mach. Intell. 4, 840–851 (2022).
Iwano, N., Adachi, T., Aoki, K., Nakamura, Y. & Hamada, M. Generative aptamer discovery using RaptGen. Nat. Comput. Sci. 2, 378–386 (2022).
Gupta, A. & Zou, J. Feedback GAN for DNA optimizes protein functions. Nat. Mach. Intell. 1, 105–111 (2019).
Linder, J. & Seelig, G. Fast activation maximization for molecular sequence design. BMC Bioinforma. 22, 510 (2021).
Bogard, N., Linder, J., Rosenberg, A. B. & Seelig, G. A deep neural network for predicting and engineering alternative polyadenylation. Cell 178, 91–106.e23 (2019).
Linder, J., Bogard, N., Rosenberg, A. B. & Seelig, G. A generative neural network for maximizing fitness and diversity of synthetic DNA and protein sequences. Cell Syst. 11, 49–62.e16 (2020).
Valeri, J. A. et al. BioAutoMATED: an end-to-end automated machine learning tool for explanation and design of biological sequences. Cell Syst. 14, 525–542.e9 (2023).
Hsu, C., Fannjiang, C. & Listgarten, J. Generative models for protein structures and sequences. Nat. Biotechnol. 42, 196–199 (2024).
Goodfellow, I. et al. Generative adversarial networks. Commun. ACM 63, 139–144 (2020).
Gulrajani, I., Ahmed, F., Arjovsky, M., Dumoulin, V. & Courville, A. C. Improved training of wasserstein GANs. In Advances in Neural Information Processing Systems (eds. Guyon, I. et al.) Vol. 30 (Curran Associates, Inc., 2017).
Cao, Y. et al. RNA-based translation activators for targeted gene upregulation. Nat. Commun. 14, 6827 (2023).
Qian, Y. et al. Programmable RNA sensing for cell monitoring and manipulation. Nature 610, 713–721 (2022).
Yan, Z. et al. Rapid, multiplexed, and enzyme-free nucleic acid detection using programmable aptamer-based RNA switches. Chem 10, 2220–2244 (2024).
Ning, H. et al. Rational design of microRNA-responsive switch for programmable translational control in mammalian cells. Nat. Commun. 14, 7193 (2023).
Kaseniit, K. E. et al. Modular, programmable RNA sensing using ADAR editing in living cells. Nat. Biotechnol. 41, 482–487 (2023).
Westbrook, A. M. & Lucks, J. B. Achieving large dynamic range control of gene expression with a compact RNA transcription–translation regulator. Nucleic Acids Res. 45, 5614–5624 (2017).
Filonov, G. S., Moon, J. D., Svensen, N. & Jaffrey, S. R. Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 136, 16299–16308 (2014).
Dunn, M. R., Jimenez, R. M. & Chaput, J. C. Analysis of aptamer discovery and technology. Nat. Rev. Chem. 1, 1–16 (2017).
Lozano, G. & Martínez-Salas, E. Structural insights into viral IRES-dependent translation mechanisms. Curr. Opin. Virol. 12, 113–120 (2015).
Sun, L. et al. Predicting dynamic cellular protein–RNA interactions by deep learning using in vivo RNA structures. Cell Res. 31, 495–516 (2021).
Zhou, Y. et al. Conditional RNA interference in mammalian cells via RNA transactivation. Nat. Commun. 15, 6855 (2024).
Zhang, H., Goodfellow, I., Metaxas, D. & Odena, A. Self-attention generative adversarial networks. In Proceedings of the 36th International Conference on Machine Learning (eds. Chaudhuri, K. & Salakhutdinov, R.) 7354–7363 (PMLR, 2019).
Riley, A. T. et al. Generative and predictive neural networks for the design of functional RNA molecules. GARDN-SANDSTORM. https://doi.org/10.5281/zenodo.15058435 (2025).
Acknowledgements
This work was supported by startup funds from Boston University; Defense Advanced Research Projects Agency (DARPA) funding (Contract No. N66001-23-2−4042); and a National Institutes of Health (NIH) U01 award (1U01AI148319-01), R01 award (1R01EB031893), and a NIH Director’s Transformative Research Award (R01EB037112) to A.A.G. A.T.R. was supported by the NIH Training Program in Quantitative Biology and Physiology (5T32GM008764). J.M.R. was supported by the NIH Training Program in Synthetic Biology and Biotechnology (1T32GM130546) and a National Science Foundation Graduate Research Fellowship (2234657). The views, opinions and/or findings expressed are those of the authors and should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. Government. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Ethics declarations
Competing interests
A.A.G. is a co-founder of En Carta Diagnostics, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Liang Huang, Hao Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article
Cite this article
Riley, A.T., Robson, J.M., Ulanova, A. et al. Generative and predictive neural networks for the design of functional RNA molecules. Nat Commun 16, 4155 (2025). https://doi.org/10.1038/s41467-025-59389-8
Received: 12 August 2023
Accepted: 16 April 2025
Published: 04 May 2025
DOI: https://doi.org/10.1038/s41467-025-59389-8

![Is the AI bubble too big to burst? [video]](https://www.youtube.com/img/desktop/supported_browsers/firefox.png)