.png)

Main
Interest in organic radical semiconductors is due to their potential applications in optoelectronic devices, biomedical technologies, quantum information systems and chiral materials1,2,3,4,5,6,7,8,9,10. Among the organic radicals family, trityl systems are of interest due to their chemical stability and photoluminescence (PL) quantum efficiency11,12. These support efficient organic light-emitting diodes (OLEDs), but are generally used as a dilute guest component in a molecular semiconductor ‘host’, since PL is generally quenched at high concentrations when inter-radical interactions are possible. Tris(2,4,6-trichlorophenyl)methyl (TTM) by itself is an alternant hydrocarbon; therefore, the energy degeneracy between the highest occupied molecular orbital (HOMO) to the singly occupied molecular orbital (SOMO) and SOMO to the lowest unoccupied molecular orbital transitions cause weak absorption and slow PL emission rates. Higher efficiencies are achieved with an attached electron donor, including carbazole and triphenylamine13,14,15, where the lowest transition is of intramolecular charge transfer (CT) from donor to TTM. PL quantum efficiencies reported for these systems that emit in the red and near-infrared regimes are often very high, particularly for red and near-infrared emission, and it has recently been shown that multiphonon decay is suppressed for these structures due to reduced vibrational coupling to the exciton16. We have also reported a wide range of TTMs with the substitution of para-chlorine with phenyl-based-substituents to adjust the colour and efficiency of emission by means of the steric bulk17,18,19. This provides a platform for red and near-infrared OLEDs.
Here we consider the effects of intermolecular interactions between radical and radical (or radical and host). Inter-radical interactions have been reported using the co-crystals of radicals and their hydrogenated radical precursors, allowing good control of inter-radical contacts. A strongly redshifted PL band at room temperature is observed above 5% radical loading, and this shows strong magnetoluminescence below 20 K (ref. 20). This is attributed to an excimer, but as noted in a recent review, the propeller shape of the TTM radical should not allow the strong π–π interactions generally required for strong excimer formation with redshifted PL, and that further work is needed to explain this redshifted emission21. We present evidence here that redshifted PL may arise from a fully charge-separated anion–cation pair, rather than from a conventional excimer. The materials explored here are shown in Fig. 1. For a regular closed-shell semiconductor, electron–hole generation requires photoexcitation across the semiconductor bandgap. By contrast, as we develop here, for radical semiconductors, electron–hole generation between neighbouring triphenyl-substituted TTMs (P3TTMs) can occur just within the SOMO non-bonding orbitals (Fig. 2a), leading to spinless anions and cations. In the absence of onsite Coulomb repulsion energies, this transfer would be barrierless. However, there is an energy cost to do this; it is the charging energy to doubly occupy the anion SOMO. This onsite Coulomb energy is the Hubbard U and this is easily measured from the voltage difference for electrochemical oxidation, where the electron is removed from the SOMO to form a cation, versus reduction in which a second electron is added to the SOMO to form an anion. It is large because the SOMO non-bonding orbitals are relatively localized. The reduction and oxidation potentials of P3TTM are –1.05 V and + 0.67 V, respectively; therefore, the energy gap is 1.72 eV corresponding to PL at 720 nm, which is close to the broad redshifted emission band18.
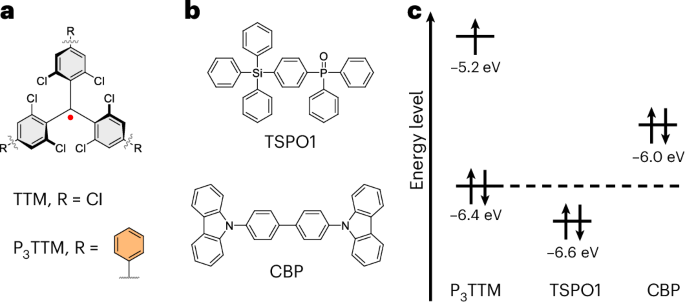
a,b, Chemical structures of TTM-based radical emitters (a) and host materials (b) investigated in this study. c, Corresponding molecular orbital energy alignment of P3TTM and host materials. The host materials only show the HOMO energy (energies are indicative and estimated from reported values18,24,25,26,27).
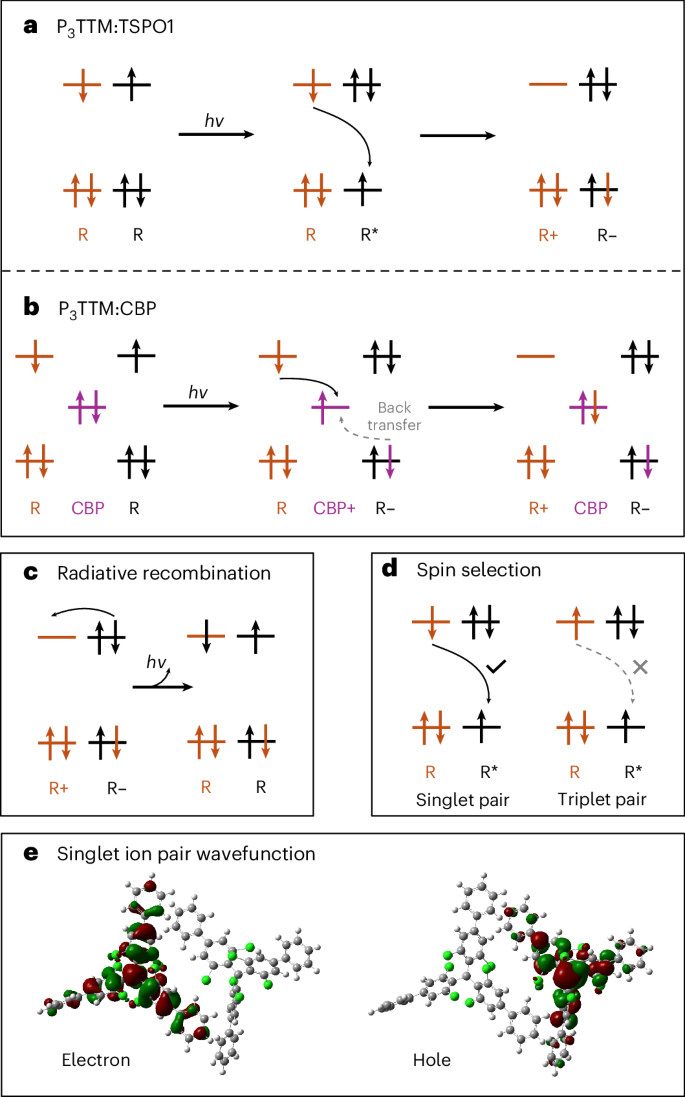
a–d, Occupancies of the single-particle states; the total ‘exciton’ energy changes that occur when the occupancies of states are changed are not shown. a, Intermolecular CT directly takes place between the ground-state radical (R) and excited-state radical (R*) to generate cations (R+) and anions (R–). b, The photogenerated hole is quickly transferred to the CBP to form R– first, followed by the second CT process between R and CBP+ that generates R+. c, Redshifted emission band is attributed to the electron–hole recombination from the ion pair. d, Magnetic field modulates the population of singlet molecular pairs, which is allowed for intermolecular CT, whereas triplet pairs are not able to form R+ and R–. e, TDDFT electron–hole wavefunction for a singlet ion pair in the P3TTM single crystal. Note how the wavefunctions with major weight on the radical centre extend over the conjugated phenyl groups.
In this study, we use P3TTM (Fig. 1a) in which the phenyl groups are in good conjugation with the TTM core (an average phenyl–phenyl dihedral angle of 34.8° is obtained from the X-ray crystal structure)18, giving PL with peak emission at 645 nm, and can provide effective inter-radical contact (Supplementary Section 8). We used a series of hole transport materials widely used in OLEDs as the host matrixes of P3TTM. These allow tuning of the host–dopant energy alignment (Fig. 1c) and allow the study of radical–radical and radical–host interactions. We also use solutions in toluene for which different concentrations can be used to control radical–radical interactions.
Photoinduced charge separation and radiative recombination in P3TTM
Radical-doped films were prepared via vacuum sublimation, as used for radical OLEDs5,13,14,22,23. In the doping range of 3 wt% to 8 wt%, differences in the observed PL are small and most results presented here are for 5 wt%. We denote films as P3TTM:host for a range of hosts used (Fig. 1b), along with an overview of the relative molecular orbital energy alignment of dopant and host materials, based on previously reported values18,24,25,26,27. We note that these hosts allow tuning of the HOMO energy gap between the dopant and hosts.
Time-resolved PL data with 400-nm excitation for P3TTM in dilute and concentrated toluene solutions and TSPO1 films are shown Fig. 3 and Supplementary Fig. 3. In the dilute solution (0.1 mM), the PL peak at 645 nm shows no spectral evolution and a mono-exponential lifetime of 9.1 ns. By contrast, the concentrated 10-mM toluene solution shows a redshifted emission band that becomes dominant at late time (around >40 ns; Fig. 3a). At this concentration, the rate of P3TTM–P3TTM collisions is high enough to ensure collision within the 9.1-ns exciton lifetime28. The associated reduction in the 645-nm PL lifetime with increased concentration follows the Stern–Volmer kinetics, indicating that this is a bimolecular process (Supplementary Section 9). Figure 3c shows the PL results for P3TTM:TSPO1. TSPO1 with its wide bandgap provides an inert environment for the radical dopants; therefore, there is no host–dopant interface effect. At early times, only the molecular emission peak at 645 nm from the molecular P3TTM exciton is observed. An additional broad emission band beyond 750 nm appears at later times. Figure 3d shows the integrated PL fraction of P3TTM:TSPO1. We see that the PL has two main contributions. At 550–750 nm, PL is mainly from the molecular exciton with a lifetime of 11.5 ns, similar to the dilute solution. However, the redshifted emission band at 750–840 nm is long-lived; around 50% of the integrated PL is detected after 500 ns. In contrast to the 645-nm emission band, which changes little ( < 2%), the redshifted emission band shows a strong magnetic field effect (MFE; Fig. 3b). The PL is suppressed at 0.7 T at room temperature and the spectrum for the change in PL intensity, ∆PL = PL (0 T) – PL (0.7 T), shows a broad band extending from 700 nm to a peak at 800 nm. We note that this MFE has been reported for ‘excimer-like’ emissions of other TTM or (3,5-dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl (PyBTM) based mono- or diradicals, and low-temperature (<20 K) magneto-PL is reported on PyBTM co-crystals20,29.
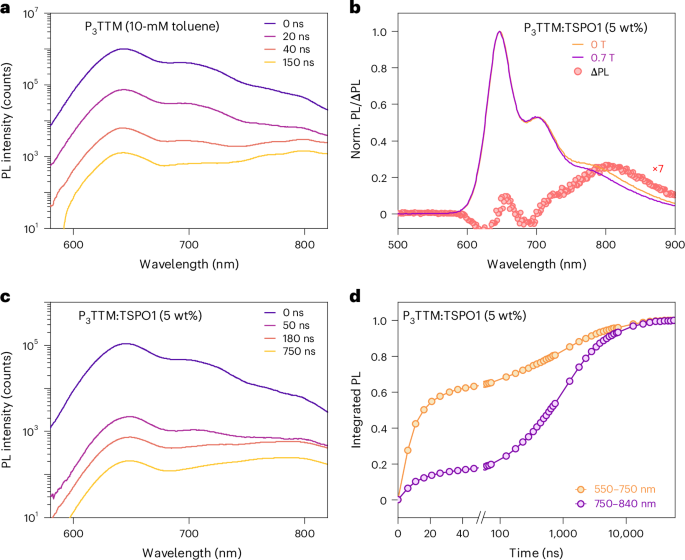
a,c, Time-resolved PL spectra of concentrated P3TTM toluene solution (10 mM; a) and P3TTM:TSPO1 (5 wt%; c). b, PL spectra of P3TTM:TSPO1 (5 wt%) under fields of 0 T and 0.7 T at room temperature and PL change under magnetic field with respect to the emission wavelength, normalized to the peak emission at 645 nm. d, Time evolution of the integrated PL fraction (obtained from the time of the PL spectra) of P3TTM:TSPO1 (5 wt%) for molecular emission and redshifted emission band.
We instead associate the redshifted emission with an inter-P3TTM CT recombination (Fig. 2c). First evidence for this comes from quantum-chemical calculations performed on close molecular pairs of the P3TTM single crystal (Supplementary Section 7). Time-dependent density functional theory (TDDFT) calculations indeed show the presence of intermolecular singlet CT states lying ~0.4 eV below the localized P3TTM excitons, in excellent agreement with the energy difference (~0.3–0.5 eV) inferred from the two main measured PL features (Fig. 3). As we hypothesized, there is substantial extension of SOMOs onto the peripheral phenyl rings (Fig. 2e) that provides the needed electronic couplings for the generation of CT pairs from the optically excited excitons and the (non-)radiative recombination of these CTs to the ground state. Applying Marcus-like rate theory to the results of TDDFT calculations performed for close pairs in the crystal yields charge generation rates up to 1 ps−1 and CT lifetimes approaching the microsecond range (Supplementary Section 7). Thus, our calculations suggest the fast formation of long-lived CT pairs that, in the absence of competing decay mechanisms, should enable efficient free charge generation, as confirmed below.
Transient optical absorption (TA) measurements are shown in Fig. 4. For a dilute toluene solution in the visible probe region (Fig. 4a), a broad photoinduced absorption (PIA) signal peaked at 675 nm and a sharp PIA in the region of 480–550 nm corresponding to the radical D1 state is probed5,12. There is no spectral shape change observed in the late time, matching the result of the time-resolved PL. However, for P3TTM:TSPO1 (Fig. 4b), two distinct PIA peaks (570–630 nm and 730–850 nm) grow, and the PIA of the radical D1 state reduces with time. We have carried out steady-state spectroelectrochemistry measurements (Fig. 4c). We observe absorptions of anions at 580 nm and cations at 760 nm and a bleaching of the ground-state absorption at 407 nm for the D0–D2 transition13. These features from spectroelectrochemistry map well with the TA features, and we associate these PIA states to closed-shell anions (570–630 nm) and cations (730–850 nm). The concentrated P3TTM solution (10 mM) shows similar long-time TA (Supplementary Figs. 9 and 10), although the CT kinetics are slower in the solution since molecular diffusion and collision need to be considered28. We note that CT can only proceed for overall spin singlet excited P3TTM (P3TTM*)–P3TTM pairs (Fig. 2d). This is consistent with the strong magneto-PL shown in Fig. 3b, and we consider that the applied magnetic field modulates the population of triplet and singlet sublevels. The reduction in singlet population with the applied field is consistent with the level crossing at the exchange energy, noting that we expect an antiferromagnetic ground state. Larger values of magnetoluminescence are reported at low temperatures and higher magnetic fields20. We emphasize that the CT between P3TTM radical pairs is very different to the standard electron–hole transfer in closed-shell semiconductors. As shown in Fig. 2a, both electrons and holes are in the SOMO levels of the two radicals, rather than the HOMO and lowest unoccupied molecular orbital with an energy gap. We note that this intermolecular CT exciton can be seen as an intermolecular analogue of the zwitterionic excited state of di- or polyradicals30,31.
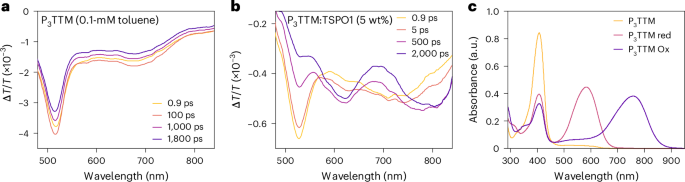
a,b, Picosecond-scale visible TA spectra of diluted P3TTM toluene solution (0.1 mM; a) and P3TTM:TSPO1 (5 wt%; b). λex = 400 nm, 17–27 μJ cm−2 per pulse. c, Spectroelectrochemistry of P3TTM in a degassed tetrahydrofuran solution, showing P3TTM, reduced P3TTM (P3TTM red) and oxidized P3TTM (P3TTM Ox).
We have previously reported a host–dopant intermolecular CT state between mesitylated TTM, M3TTM and a donor CBP32 host (structure shown in Fig. 1), which gives long-lived emission, where the TTM acts as an electron acceptor17. Figure 5 shows the effects of CBP host matrixes on intermolecular CT. The PL of a P3TTM:CBP system is shown in Fig. 5a, exhibiting similar behaviour as the P3TTM:TSPO1 system, with a magnetic-field-dependent redshifted band centred near 790 nm, which is also time delayed (Supplementary Fig. 5). This indicates broadly similar behaviour as the P3TTM:TSPO1 system, but as we explore below, there is an intermediate time regime in which CBP has transferred an electron to the P3TTM. This can then be followed by either back transfer from the P3TTM anion (causing a longer molecular emission lifetime of 13.7 ns; Supplementary Fig. 8) and subsequent electron transfer to CBP from a neutral P3TTM.
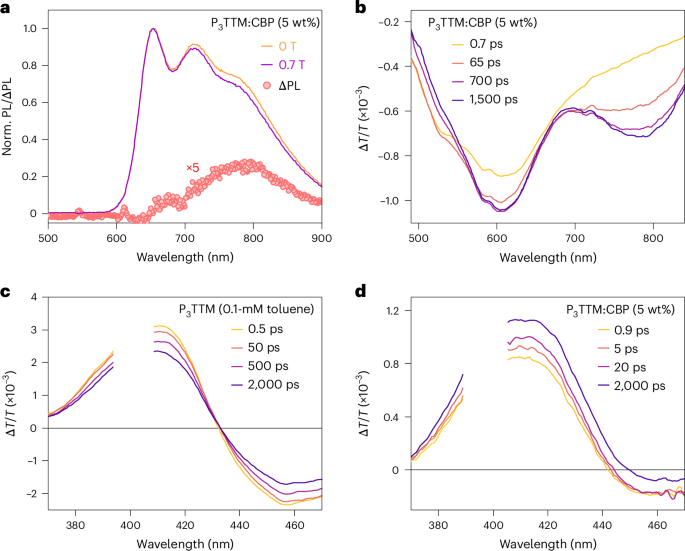
a, PL spectra of P3TTM:CBP (5 wt%) under fields of 0 T and 0.7 T at room temperature and PL change under magnetic field with respect to the emission wavelength, normalized to the emission at 645 nm. b, Visible TA spectrum of P3TTM:CBP. c,d, Ultraviolet TA spectra of diluted P3TTM toluene solution (0.1 mM; c) and P3TTM:CBP (5 wt%; d). The spectrum break is due to pump laser scattering (λex = 400 nm, 12–13 μJ cm−2 per pulse).
The visible TA spectrum shown in Fig. 5b of the CBP film has two similar broad PIA peaks in the same region of 570–630 nm and 730–850 nm as the TSPO1 film, as the same radical anions and cations are generated, but the 730–850-nm band (associated with the P3TTM cation) grows in more slowly over 100 ps. Given the 5% P3TTM fraction, the CT process most accessible will be from CBP to P3TTM, and we attribute the slower build-up of the P3TTM cation population to the subsequent CT to bring CBP back to zero charge. Access to the CBP–P3TTM CT process allows a faster initial growth of the P3TTM anion population compared with the P3TTM:TSPO1 system. We also note that the first CT process between CBP and P3TTM* is not spin selective; spinless CBP–P3TTM* pairs give quicker CT kinetics, but the subsequent CT between positively charged CBP, CBP+ and P3TTM is dependent on the spin state of the CBP+–P3TTM pairs. Therefore, the PL of P3TTM:CBP also has an MFE for the redshifted emission band (Fig. 5a). In both TSPO1 and CBP, the growth rate of the anion and cation PIA increases for a higher doping concentration or closer contact between radicals. The CT population build-up times are shortened to several picoseconds in the 100-wt% doped film (without hosts) due to a short inter-radical distance (Supplementary Figs. 30 and 31).
TA of the ground-state bleach (GSB) for P3TTM (360–430 nm) is shown for dilute solutions in Fig. 5c and for P3TTM:CBP films in Fig. 5d. For the dilute solution, GSB is the largest at early times and its later decay is consistent with the 9.1-ns PL decay. By contrast, the P3TTM:CBP film shows GSB growth up to 2 ns. The early time GSB is due to the P3TTM exciton evolving into P3TTM anions, accompanied by CBP cations. The GSB growth is due to the conversion of CBP cations to P3TTM cations as the full P3TTM anion–cation population develops. In the first 2 ns, although the GSB for the dilute solution falls by 27%, for the P3TTM:CBP film, the GSB rises by 32%. This indicates that the quantum yield for charge photogeneration is remarkably high, which we estimate to be up to 40% (Supplementary Section 5).
Direct-charge photogeneration
Our studies of P3TTM in solution and in solid hosts indicate clear evidence for a fully charge-separated anion–cation intermolecular state, which shows PL near 800 nm. Excitation fluence measurements (Supplementary Fig. 7) indicate that this anion–cation pair is mostly bound at room temperature. We have investigated solid films of P3TTM without a host material in standard diode device structures, showing that it is possible to fully separate electrons and holes with an applied field. The films show substantially reduced PL (Supplementary Table 4) and tracked through reduced photoexcited-state lifetimes (Supplementary Figs. 30 and 31). The TA spectra shown in Supplementary Fig. 32 reveal clear evidence for early time charge separation, with anion (600 nm) and cation (800 nm) bands fully formed by 4 ps, but rapid decay to the ground state within a few nanoseconds. In spite of these reduced lifetimes, we are still able to get long-range charge separation in biased diode structures.
We fabricated standard multilayer diode structures using poly(3,4-ethylene dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) on indium tin oxide (ITO) as the hole injection/extraction electrode, and fullerene (C60)/bathocuproine (BCP)/aluminium (Al) as the electron injection/extraction layers. These were selected to give band alignment with the P3TTM radical. We also made devices using rubrene as a control, since this has similar redox potentials. The device architecture used is shown in Fig. 6a for ITO (150 nm)/PEDOT:PSS (40 nm)/photoactive layer (P3TTM or rubrene) (80 nm)/C60 (20 nm)/BCP (5 nm)/Al (100 nm). Figure 6b shows the photocurrent and dark current under bias for both P3TTM and rubrene devices (both made with and without the C60 layer and photoexcitation at 395 nm was 160 mW cm−2).
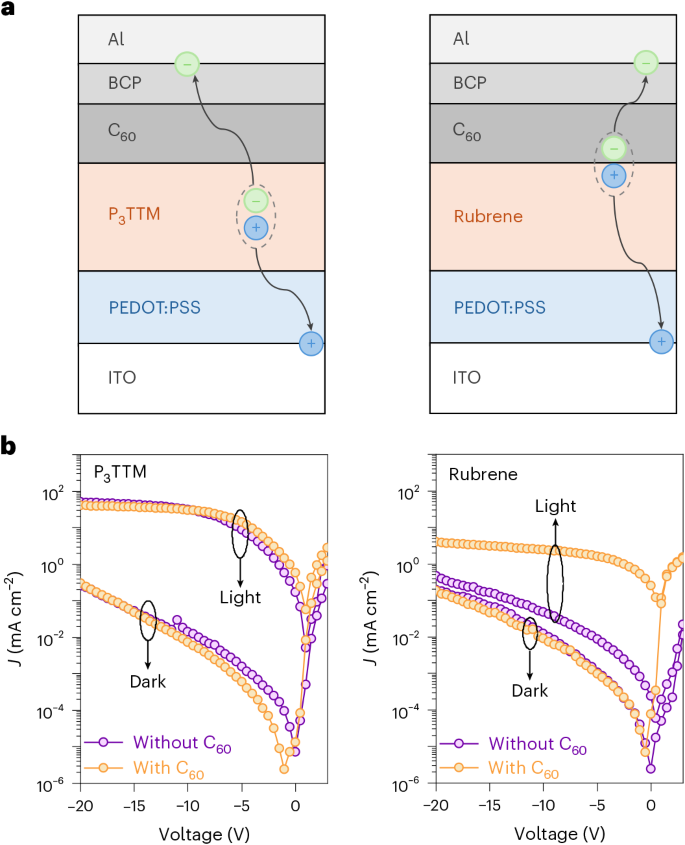
a, Device architecture and schematic of the charge separation process in the P3TTM device (left) and rubrene device (right). b, Photocurrent density under 395-nm excitation at 160 mW cm−2 and dark current density (J) comparison of the P3TTM device (left) and rubrene device (right).
The rubrene device shows expected behaviour for charge photogeneration at the rubrene/C60 heterojunction, giving a short-circuit current density of 0.25 mA cm−2 and a quantum yield of 4.9%, considering 90% transmission rate of the rubrene thin film under ultraviolet (UV) light. This is consistent with charge photogeneration within an exciton diffusion range of 6–8 nm in rubrene33. Under reverse bias, these devices showed little current increase, rising to 4 mA cm−2 at –20 V. For diodes made without the C60 layer, the photocurrents are much lower, similar in magnitude to the dark current, showing that the rubrene/BCP heterojunction does not generate free charge carriers.
By contrast, the P3TTM diode showed a low quantum yield at short circuit, but a very strong increase in reverse bias that saturates at around 45 mA cm−2, indicating a quantum yield for charge collection close to 100% and considering a 15% transmission rate of the P3TTM thin film under UV light. The behaviour was very similar for devices made with and without the C60 layer, indicating that charge photogeneration was not controlled by the P3TTM/C60 interface. Rather, this close-to-unity charge collection efficiency is due to bulk charge photogeneration, as described above.
The ability to fully separate electrons and holes following photoexcitation in a diode structure made with a single non-polar semiconductor is routine in mainstream inorganic semiconductors such as silicon, in which the photogenerated exciton is large and has low binding energy (<10 meV), but for molecular systems, this has rarely been previously reported. We note that there are interesting studies on ‘symmetry-breaking’ CT at an intramolecular level, often in the presence of a polar solvent34,35,36,37,38,39. We also note that our demonstrated high-efficiency of charge photogeneration precludes any ‘excimer’ formation, which typically do not undergo charge separation.
Outlook
In summary, the intermolecular interaction and excited-state dynamics of neutral π radicals are studied by various time-resolved techniques and steady-state spectroscopies. In both solutions and films, we identified an intermolecular symmetry-breaking charge separation process between P3TTM SOMOs to form an excited-ion pair state comprising a pair of closed-shell anion and cation. The GSB growth of P3TTM provides further evidence for the intermolecular process. We also demonstrated that the redshifted PL has an MFE, as the intermolecular CT depends on the spin state of P3TTM*–P3TTM and CBP+–P3TTM intermediates. This mechanism can be generalized to other open-shell materials, for example, the long-lived and redshifted emission generally observed for TTM- and PyBTM-based radicals can be attributed to the recombination of this CT exciton. We also explored the effect of host–dopant interface on the intermolecular CT channels. This work demonstrates that the photoinduced charge separation can be intermolecularly driven in neutral radicals and that the unpaired electron can be mobile and not bound to the radical centre. Homojunction charge separation, as observed here, has been a long-sought-after goal in organic photovoltaics40. This work provides an avenue for the exploration of power generation and solar-driven chemistry in both solution and solid state using only a single component.
Methods
Film preparation
Radicals were synthesized as previously reported18. Host materials were obtained from Ossila without further purification. Thin films were prepared by thermal evaporation under vacuum (~10−7 torr, Angstrom Engineering EvoVac 700 system). Here 100 nm of 5 wt% radical-doped CBP, TSPO1 and TAPC films were deposited on UV fused silica substrates. Polymethyl methacrylate films were prepared by spin coating in a nitrogen-filled glovebox. All the films were encapsulated in this glovebox. The doping concentration stated in this study denotes the weight percentage.
Device fabrication and characterization
Organic transport layer materials were obtained from Ossila without further purification. The ITO substrate was cleaned in an ultrasonic bath of detergent, deionized water, acetone and isopropanol for 10 min each. It was then UV treated in a UV–ozone chamber for 12 min. A thin layer of PEDOT:PSS (Clevios P VP AI 4083) was prepared by spin coating the PEDOT:PSS solution at 3,000 rpm for 40 s on the ITO substrate and annealed at 150 °C for 15 min in air. Layers of P3TTM, C60, BCP and Al were deposited by thermal evaporation under a vacuum (~10−7 torr, Angstrom Engineering EvoVac 700 system) at a rate of 0.1–1 Å s−1. The whole device has a structure of ITO (150 nm)/PEDOT:PSS (40 nm)/P3TTM (80 nm)/C60 (20 nm)/BCP (5 nm)/Al (100 nm). A Keithley 2635A source with a 395-nm light source was used to measure the current–voltage plot. The voltage was applied to the device between −20 V and 3 V at a sweep speed of 0.5 V s−1. The effective electrode overlap area was 4.5 mm2, which was used to calculate the current density.
Steady-state photophysics measurements
The absorption spectra were measured by a commercially available Shimadzu UV-2550 spectrophotometer and a Shimadzu UV-1800 spectrophotometer. PL was measured by a custom-made setup by providing continuous photoexcitation at 405 nm from a laser diode. The PL from the samples was collected in a collimating two-lens apparatus and directed into an optical fibre that supplies photons into a calibrated grating spectrometer (Andor SR-303i) and finally into a silicon camera where it is recorded.
Transient PL spectroscopy
Transient PL (time-resolved PL) spectra at nanosecond–microsecond timescales were recorded using an electronically gated intensified charge-coupled device camera (Andor iStar DH740 CCI-010) connected to a calibrated grating spectrometer (Andor SR-303i). A narrowband non-colinear optical parametric amplifier pumped with a frequency-doubled output of a 1-kHz 800-nm laser pulse (100-fs duration) from a Ti:sapphire amplifier (Spectra Physics Solstice Ace) was used to generate a tuneable excitation pulse. A 400-nm excitation can be achieved by the second harmonic of the 800-nm output, generated using a β-barium borate crystal. Here 425-nm long-pass filters (Edmund Optics) were used to prevent scattered laser signals from entering the spectrometer. Temporal evolution of the emission was obtained by stepping the intensified charge-coupled device delay with respect to the excitation pulse, with a minimum gate width of 5 ns. Recorded data were corrected to account for filter transmission and camera sensitivity.
TA spectroscopy
The picosecond-scale TA spectroscopies were carried out either on a custom-made TA setup or a commercialized TA setup. The custom-made TA setup has an output from a Ti:sapphire amplifier (Spectra Physics Solstice Ace) that generated 100-fs-duration pulses centred at 800 nm with a 1-kHz repetition rate. The pulse was split into pump and probe beams. The 400-nm excitation pump was generated by passing the second harmonic of the 800-nm output using a β-barium borate crystal. The pump light was chopped at 500 Hz by a chopper wheel. The visible probe light was generated via non-collinear optical parametric amplifiers. The pump–probe time delay was provided by a mechanical delay stage (Thorlabs DDS300-E/M). The probe pulses were split into two beams by a 50/50 beamsplitter to provide a reference beam, which increases the signal-to-noise ratio. The probe pulses were detected by a silicon (Hamamatsu S8381-1024Q) dual-line array with a custom-built board from Stresing Entwicklungsbüro. The fundamental laser beam of the commercialized picosecond-scale TA setup at 1,030 nm was provided by PHAROS. The ORPHEUS system, which was pumped by an ytterbium-doped solid-state chirped pulse amplifier, generated pump lights using 90% of the fundamental laser (LIGHT CONVERSION). The pump wavelength can be varied from 350 nm to 2,000 nm with a 10-kHz repetition rate and an ~100-fs pulse duration. The rest of the 10% fundamental beam was injected into HARPIA-TA to generate the probe light. The pump–probe time delay was provided by a mechanical delay stage. The probe pulses were detected by a spectrograph with dual outputs to cover the UV–near-infrared regions (Kymera-193i-B2).
Magneto-PL measurement
The encapsulated film sample was positioned between magnet cores (GMW 3470 electromagnet). Its PL spectrum was recorded with an Andor SR-303i spectrometer with and without a magnetic field. The magnetic field was swept from 0 T to 0.7 T with a ramping step of 0.01 T at room temperature. The continuous photoexcitation at 405 nm was provided by a laser diode.
Spectroelectrochemistry
A commercially available PalmSens EmStat4S potentiostat was connected to a commercially available Shimadzu UV-1800 spectrophotometer. The measurements were carried out in a custom-made three-electrode setup using a quartz cuvette (0.1-mm path length) as the spectroelectrochemical cell, a coil of platinum wire as the working electrode (in the light path), a platinum wire as the counter electrode and a freshly activated silver wire as the Ag/Ag+ reference electrode. The silver wire was activated by immersing in a concentrated HCl solution to remove any silver oxides or other impurities, then rinsed with water and acetone and dried before measurements. The reference electrode was calibrated against ferrocene/ferrocenium (Fc/Fc+) redox couple. For this setup, Fc/Fc+ half-wave potential (E1/2) was determined at 0.40 V versus Ag/Ag+. The supporting electrolyte was 0.1-M solution of Bu4NPF6 in anhydrous tetrahydrofuran. The electrolyte was bubbled with Ar gas before measurements to remove any dissolved oxygen and the measurements were carried out under an Ar atmosphere. The sample concentration was adjusted to keep the optical density below 1.0 a.u. for neutral radicals. The absorption spectra of the oxidized (cationic) and reduced (anionic) species were measured by applying a constant potential of +1.1 V and –1.4 V versus Fc and Fc+, respectively. These are ~0.4 V higher than the corresponding half-wave potentials determined for the P3TTM radical18.
Quantum-chemical calculations
Molecular pairs in close contact selected from the X-ray crystal structure of P3TTM were used for the calculation of the excited-state properties (vertical excitations, transition dipole moments and state dipoles) by means of TDDFT within the Tamm–Dancoff approximation41, in conjunction with the LC-ωhPBE functional and the 6-311G(d,p) basis set42. Furthermore, to implicitly take into account dielectric screening effects, the screened range-separated hybrid procedure was applied43. In such a scheme, the range-separation parameter ω was set to 0.100 Bohr−1, whereas the other parameters (α and β) were modified according to the dielectric constant of the solvent/environment. In this work, we chose a dielectric constant typical of toluene, that is, ε = 2.37. Regarding the electronic coupling calculations and the (non-)radiative charge dissociation/recombination rates (Supplementary Information), all the calculations were performed using the Gaussian 16 package.
Data availability
The optical spectroscopy and device characterization data generated in this study have been deposited in the University of Cambridge Repository at https://doi.org/10.17863/CAM.120958. Source data are provided with this paper.
References
Peng, Q., Obolda, A., Zhang, M. & Li, F. Organic light-emitting diodes using a neutral p radical as emitter: the emission from a doublet. Angew. Chem. Int. Ed. 54, 7091–7095 (2015).
Ji, L., Shi, J., Wei, J., Yu, T. & Huang, W. Air-stable organic radicals: new-generation materials for flexible electronics?. Adv. Mater. 32, 1908015 (2020).
Cui, X. et al. Stable π-radical nanoparticles as versatile photosensitizers for effective hypoxia-overcoming photodynamic therapy. Mater. Horiz. 8, 571–576 (2021).
Gorgon, S. et al. Reversible spin-optical interface in luminescent organic radicals. Nature 620, 538–544 (2023).
Ai, X. et al. Efficient radical-based light-emitting diodes with doublet emission. Nature 563, 536–540 (2018).
Mayorga Burrezo, P. et al. Organic free radicals as circularly polarized luminescence emitters. Angew. Chem. Int. Ed. 58, 16282–16288 (2019).
Mas-Torrent, M. et al. Organic radicals on surfaces: towards molecular spintronics. J. Mater. Chem. 19, 1691–1695 (2009).
Wu, S. et al. Toward two-dimensional π-conjugated covalent organic radical frameworks. Angew. Chem. Int. Ed. 57, 8007–8011 (2018).
Gu, Q., Abdurahman, A., Friend, R. H. & Li, F. Polymer light emitting diodes with doublet emission. J. Phys. Chem. Lett. 11, 5638–5642 (2020).
Teki, Y. Excited-state dynamics of non-luminescent and luminescent π-radicals. Chem. Eur. J. 26, 980–996 (2020).
Murto, P. & Bronstein, H. Electro-optical π-radicals: design advances, applications and future perspectives. J. Mater. Chem. C 10, 7368–7403 (2022).
Guo, H. et al. High stability and luminescence efficiency in donor–acceptor neutral radicals not following the Aufbau principle. Nat. Mater. 18, 977–984 (2019).
Abdurahman, A. et al. Understanding the luminescent nature of organic radicals for efficient doublet emitters and pure-red light-emitting diodes. Nat. Mater. 19, 1224–1229 (2020).
Cho, H.-H. et al. Efficient near-infrared organic light-emitting diodes with emission from spin doublet excitons. Nat. Photon. 18, 905–912 (2024).
Lu, C. et al. Achieving nearly 100% photoluminescence quantum efficiency in organic radical emitters by fine-tuning the effective donor-acceptor distance. Adv. Funct. Mater. 34, 2314811 (2024).
Ghosh, P. et al. Decoupling excitons from high-frequency vibrations in organic molecules. Nature 629, 355–362 (2024).
Murto, P. et al. Mesitylated trityl radicals, a platform for doublet emission: symmetry breaking, charge-transfer states and conjugated polymers. Nat. Commun. 14, 4147 (2023).
Murto, P. et al. Steric control of luminescence in phenyl-substituted trityl radicals. J. Am. Chem. Soc. 146, 13133–13141 (2024).
Ohisa, S. & Honda, S. Luminescence enhancement by symmetry-breaking in the excited state in radical organic light-emitting diodes. Commun. Chem. 6, 238 (2023).
Kimura, S. et al. Magnetoluminescence in a photostable, brightly luminescent organic radical in a rigid environment. Angew. Chem. Int. Ed. 57, 12727–12731 (2018).
Mizuno, A., Matsuoka, R., Mibu, T. & Kusamoto, T. Luminescent radicals. Chem. Rev. 124, 1034–1121 (2024).
Gu, Q. et al. Fast transfer of triplet to doublet excitons from organometallic host to organic radical semiconductors. Adv. Mater. 36, 2402790 (2024).
Cho, H. H. et al. Near-infrared light-emitting diodes from organic radicals with charge control. Adv. Opt. Mater. 10, 2200628 (2022).
Yadav, R. A. K., Dubey, D. K., Chen, S.-Z., Liang, T.-W. & Jou, J.-H. Role of molecular orbital energy levels in OLED performance. Sci. Rep. 10, 9915 (2020).
Vasilopoulou, M. et al. High efficiency blue organic light-emitting diodes with below-bandgap electroluminescence. Nat. Commun. 12, 4868 (2021).
Kim, B. S. & Lee, J. Y. Engineering of mixed host for high external quantum efficiency above 25% in green thermally activated delayed fluorescence device. Adv. Funct. Mater. 24, 3970–3977 (2014).
Cho, H. H. et al. Efficient and bright organic radical light-emitting diodes with low efficiency roll-off. Adv. Mater. 35, 2303666 (2023).
Walker, B. J., Musser, A. J., Beljonne, D. & Friend, R. H. Singlet exciton fission in solution. Nat. Chem. 5, 1019–1024 (2013).
Kimura, S. et al. A ground-state-dominated magnetic field effect on the luminescence of stable organic radicals. Chem. Sci. 12, 2025–2029 (2021).
Yu, C. P. et al. Near-infrared luminescent open-shell π-conjugated systems with a bright lowest-energy zwitterionic singlet excited state. Sci. Adv. 10, eado3476 (2024).
Zhou, Z. et al. Sulfone-functionalized Chichibabin’s hydrocarbons: stable diradicaloids with symmetry breaking charge transfer contributing to NIR emission beyond 900 nm. J. Am. Chem. Soc. 146, 6763–6772 (2024).
Guo, J. et al. Novel triazine derivatives with deep LUMO energy levels as the electron-accepting components of exciplexes. J. Mater. Chem. C 9, 939–946 (2021).
Halls, J. J., Pichler, K., Friend, R. H., Moratti, S. & Holmes, A. Exciton diffusion and dissociation in a poly(p-phenylenevinylene)/C60 heterojunction photovoltaic cell. Appl. Phys. Lett. 68, 3120–3122 (1996).
Kaul, N. & Lomoth, R. The carbene cannibal: photoinduced symmetry-breaking charge separation in an Fe(III) N-heterocyclic carbene. J. Am. Chem. Soc. 143, 10816–10821 (2021).
Ramirez, C. E. et al. Symmetry-breaking charge separation in the solid state: tetra(phenoxy)perylenediimide polycrystalline films. J. Am. Chem. Soc. 142, 18243–18250 (2020).
Wega, J. & Vauthey, E. Bimolecular photoinduced symmetry-breaking charge separation of perylene in solution. Photochem. Photobiol. Sci. 23, 93–105 (2024).
Markovic, V., Villamaina, D., Barabanov, I., Lawson Daku, L. M. & Vauthey, E. Photoinduced symmetry-breaking charge separation: the direction of the charge transfer. Angew. Chem. Int. Ed. 50, 7596–7598 (2011).
Whited, M. T. et al. Symmetry-breaking intramolecular charge transfer in the excited state of meso-linked BODIPY dyads. Chem. Commun. 48, 284–286 (2012).
Trinh, C. et al. Symmetry-breaking charge transfer of visible light absorbing systems: zinc dipyrrins. J. Phys. Chem. C 118, 21834–21845 (2014).
Dong, Y. et al. Orientation dependent molecular electrostatics drives efficient charge generation in homojunction organic solar cells. Nat. Commun. 11, 4617 (2020).
Hirata, S. & Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 314, 291–299 (1999).
Frisch, M. J. et al. Gaussian 16, Revision A.03 (Gaussian, Inc., 2016).
Refaely-Abramson, S. et al. Gap renormalization of molecular crystals from density-functional theory. Phys. Rev. B 88, 081204 (2013).
Acknowledgements
We thank A. Bond for carrying out the X-ray crystallography measurements and data analysis at the Yusuf Hamied Department of Chemistry, University of Cambridge. P.M. and R.C. have received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement numbers 891167 and 859752. R.H.F., B.L., R.C. and P.M. acknowledge funding from the European Research Council under the European Union’s Horizon 2020 research and innovation programme grant agreement number 101020167. P.M. acknowledges funding from the Research Council of Finland (No. 363345). G.L. acknowledges the Italian Ministry of University and Research for funding provided by the European Union-NextGenerationEU-PNRR, Missione 4, Componente 2, Linea di investimento 1.2. Computational resources in Mons were provided by the FNRS ‘Consortium des Equipements de Calcul Intensif-CECI’ program grant number 2.5020.11. D.B. is the Research Director of the Belgian National Fund for Scientific Research (FRS-FNRS).
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Materials thanks Christopher Bardeen, Kaifeng Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Li, B., Murto, P., Chowdhury, R. et al. Intrinsic intermolecular photoinduced charge separation in organic radical semiconductors. Nat. Mater. (2025). https://doi.org/10.1038/s41563-025-02362-z
Received: 31 October 2024
Accepted: 01 September 2025
Published: 30 September 2025
DOI: https://doi.org/10.1038/s41563-025-02362-z