.png)

Researchers conduct chemistry simulations with sample-based quantum diagonalization (SQD), a prime candidate for near-term demonstrations of quantum advantage.
In a new paper published in The Journal of Chemical Theory and Computation, researchers from IBM Quantum® and Lockheed Martin demonstrate how a quantum computer can help accurately model the electronic structure of certain molecules. These so-called, “open-shell” molecules contain one or more unpaired electrons, making them difficult to simulate with classical methods alone.
The new research marks the first application of the sample-based quantum diagonalization (SQD) technique to open-shell systems — an important milestone for quantum chemistry and its applications in aerospace, sensing, and materials design. IBM® researchers believe that SQD is a prime candidate for near-term demonstrations of quantum advantage, as it allows researchers to combine the best of high-performance quantum computers and high-performance classical computers in tackling interesting simulation problems.
Quantum computing for chemistry: Why now?
Quantum chemistry has long stood out as one of the most promising applications for quantum computing. Many chemical systems — particularly those involving strong electron correlation, e.g., transition metals, radical species, transition states, or excited states — are exceptionally hard to simulate using classical high-performance computing.
The computational cost of exactly modeling these systems with classical computers grows exponentially with the number of interacting electrons, making exact solutions practically inaccessible for even relatively small molecules. While there are some classical approximation methods that can simulate chemical systems with strong electronic correlation, these methods are computationally expensive.
This is where quantum computers could come in. Quantum chemistry simulations run on quantum computers can accurately compute the electronic structure and energies of these systems as an alternative to classical methods. As quantum computers progress toward fault-tolerance, it is possible that they will be able to simulate strongly correlated systems more accurately and at much larger scales than any classical approximation method.
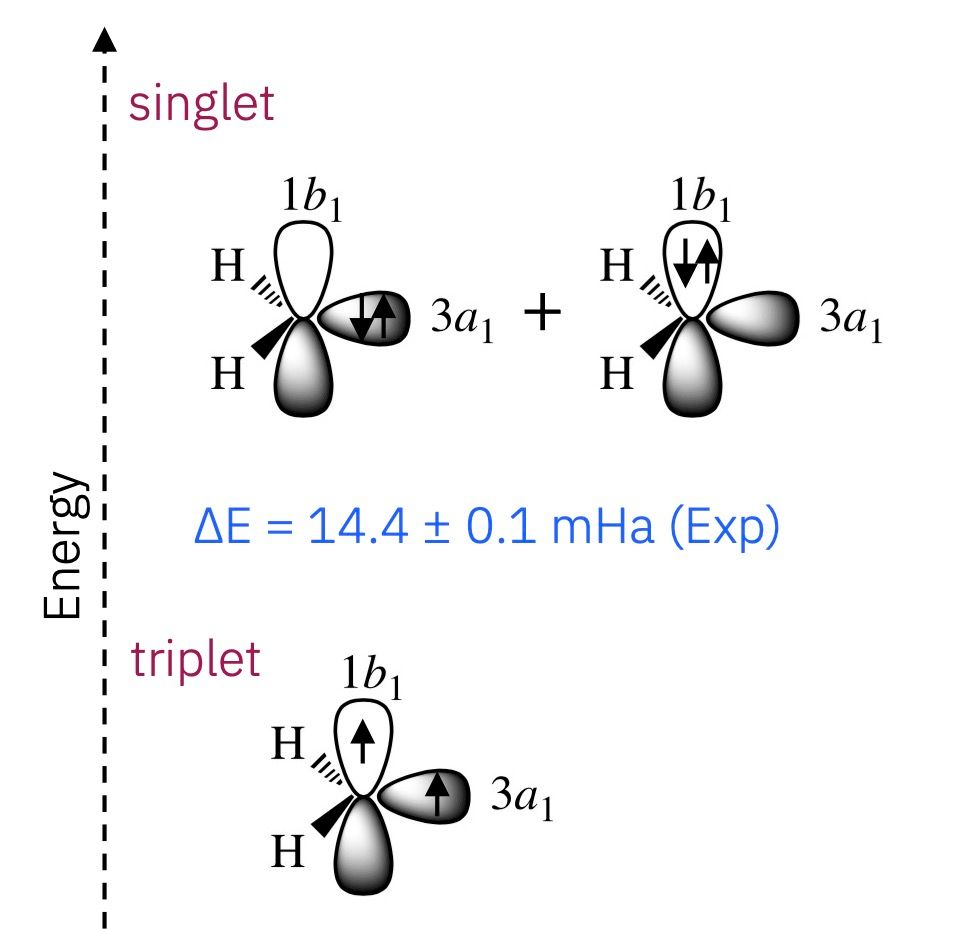
One especially important quantity in computational chemistry is the energy difference between the electronic states of a molecule, such as the gap between a singlet and a triplet state — more on those later. These electronic transitions play a central role in everything from chemical reactivity and photochemistry to material properties and molecular sensing.
Accurately computing transition energies enables scientists to:
- Predict reactivity and mechanisms in catalytic and combustion reactions
- Design molecules with tailored optical or electronic properties, such as fluorescent probes or solar absorbers
- Model excited-state behavior in materials used for sensing, display technologies, or aerospace applications
- Benchmark and validate experiments in cases where measurements are difficult, dangerous, or expensive
But accurate calculation of transition energies is also extremely challenging. As molecules become more complex, the number of possible interactions between electrons (i.e., the electron correlation) grows exponentially. Capturing these interactions accurately, especially when electrons are strongly correlated or unpaired (as in radical species), pushes classical computing methods to their limits.
Things become even more challenging when we’re dealing with open-shell molecules, i.e., molecules that have unpaired electrons in their electronic configuration. Closed-shell molecules tend to have relatively simple, stable wavefunctions because all electrons are paired in orbitals. Open-shell systems are more complex. They can exhibit magnetic properties, high reactivity, and complex “multi-reference” electronic structures where we must rely on multiple wave functions to capture the full complexity. These characteristics make open-shell systems incredibly important in fields like combustion chemistry, catalysis, and atmospheric science — but also make them difficult to model accurately.
Open-shell species include radicals, transition metal complexes, and intermediate states in chemical reactions. Because their unpaired electrons create a rich but difficult-to-capture correlation structure, traditional classical methods often struggle with them — either oversimplifying the physics or requiring extensive computational resources.
This is precisely where quantum computing shows promise. The ability of quantum algorithms to directly encode and process the electron entanglement gives them an edge for tackling open-shell systems. When integrated into IBM’s quantum-centric supercomputing architecture — which combines quantum processors with powerful classical resources — these methods can help researchers simulate open-shell molecules with higher accuracy and scalability than ever before.
Ultimately, this progress supports a wide range of applications, from understanding reaction mechanisms in combustion engines to designing novel materials and molecular sensors. And it’s exactly this type of challenge that Lockheed Martin and IBM set out to address with the recent CH2 study.
Why methylene matters and key results
Methylene, or CH2, has only three atoms. However, while it might appear to be a simple molecule, it plays a key role in combustion emissions, atmospheric chemistry, and interstellar processes due to its high reactivity, and it is essential in chain reactions in combustion and organic chemistry.
From an electronic structure perspective, CH2 is a molecule composed of one carbon atom and two hydrogen atoms. In its ground state, CH2 is a diradical that adopts a triplet electronic structure configuration, where the carbon atom's outer shell contains two unpaired electrons which have parallel spins, leading to a total spin of one.
This configuration is typically energetically high, and generally not found in ground states. The unpaired electrons give the molecule magnetic properties compared to more stable, closed-shell molecules. They also contribute to Methylene’s high reactivity, making it a crucial intermediate in combustion reactions, where bonds break and form rapidly as molecules are consumed in flames or engines. An intermediate is an unstable molecule that is temporarily generated and then quickly consumed in a multi-step chemical reaction.
The first excited state CH2 is called a carbene singlet state, where the two electrons in the carbon atom are paired and one orbital is empty. In a singlet state, the two electrons are paired with opposite spins, resulting in a total spin of zero. This is usually the lower-energy, more stable configuration. However, that’s not the case for CH2, which is a relatively rare example of a molecule whose triplet state is lower energy than its singlet state. In the triplet state, the two electrons are unpaired with spins pointing in the same direction.
The energy difference between these states is known as the singlet-triplet energy gap, and accurately predicting this gap is essential for understanding how CH2 interacts in complex chemical processes.
In this study, the quantum simulation of the electronic structure of CH2 —specifically its singlet and triplet states — was critical because traditional classical computational chemistry methods struggle to provide accurate results for open-shell molecules. Complicating matters further was the fact that the Lockheed Martin and IBM researchers set out to compare CH2 to the carbene structure.
This makes quantum computers, with their ability to naturally handle electron correlations and complex wavefunctions, an ideal tool for simulating the reactivity of CH2 and other radical species. The results from quantum simulations offer insights into the molecule’s dissociation energy, energy gap between states, and reactivity in ways that classical models cannot fully capture.
By accurately modeling the dissociation of the C—H bond in CH2 and the electronic transitions between singlet and triplet states, we can improve our understanding of how such species behave in real-world conditions — enabling more reliable modeling of combustion emissions and contributing to advancements in sensor technology. These molecules emerge during bond-breaking reactions, and accurately predicting their behavior is vital for developing better models of combustion emissions, propulsion systems, and even chemical sensing technologies.
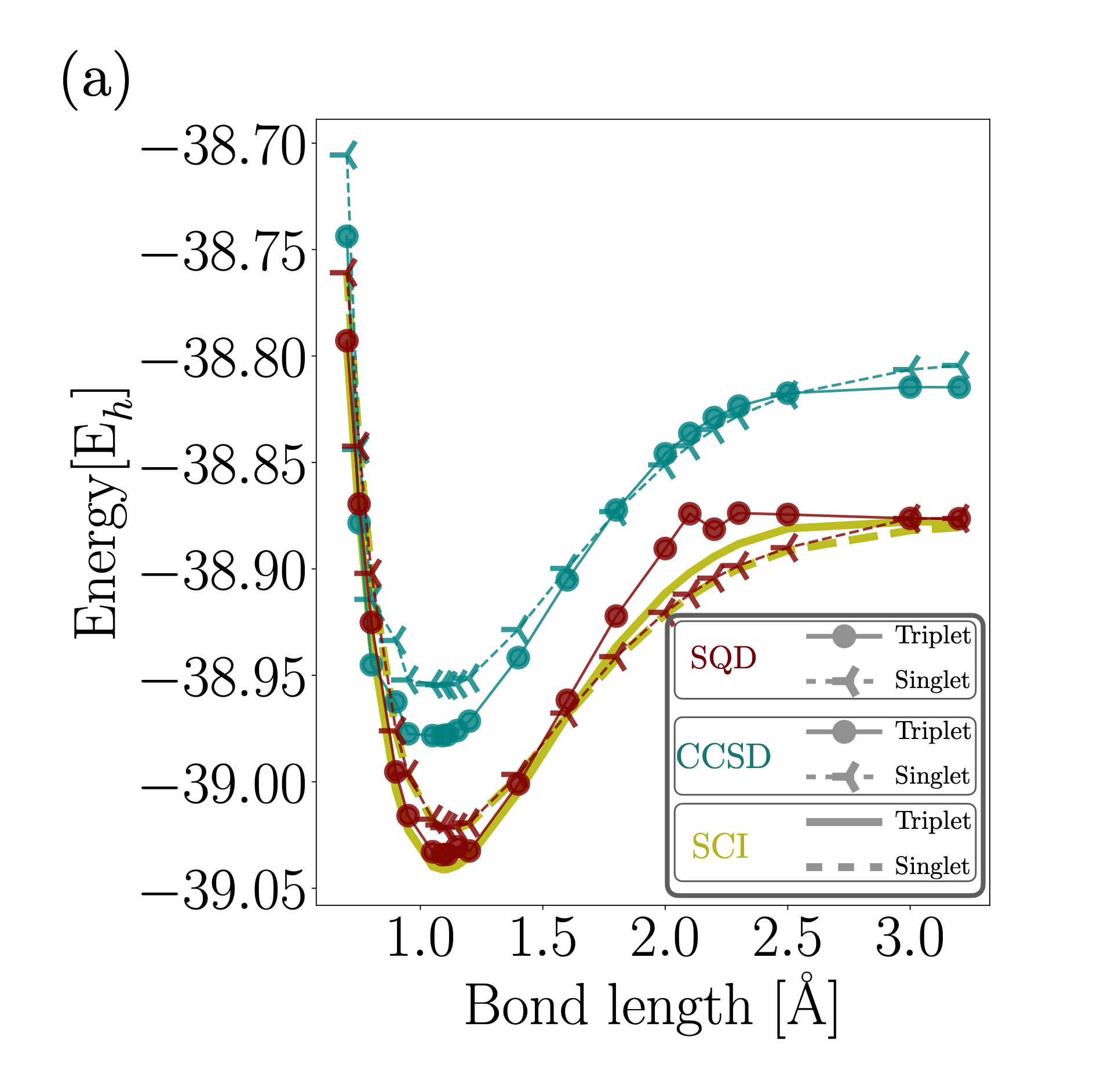
Using SQD on a quantum processor, the team successfully calculated both the singlet and triplet states, including their dissociation energies and energy gaps. The results were benchmarked against high-accuracy classical methods (Selected Configuration Interaction, or SCI), showing:
- Strong agreement for singlet dissociation energy, within a few milli Hartrees of the SCI reference.
- Consistent triplet energies near equilibrium geometries.
- Accurate prediction of the singlet-triplet energy gap, aligning with both experiment and classical simulation.
Crucially, this is the first-ever application of the SQD method to an open-shell system, establishing a new level of credibility for the ability of quantum methods in tackling molecules with complex electronic structures like radicals and carbenes.
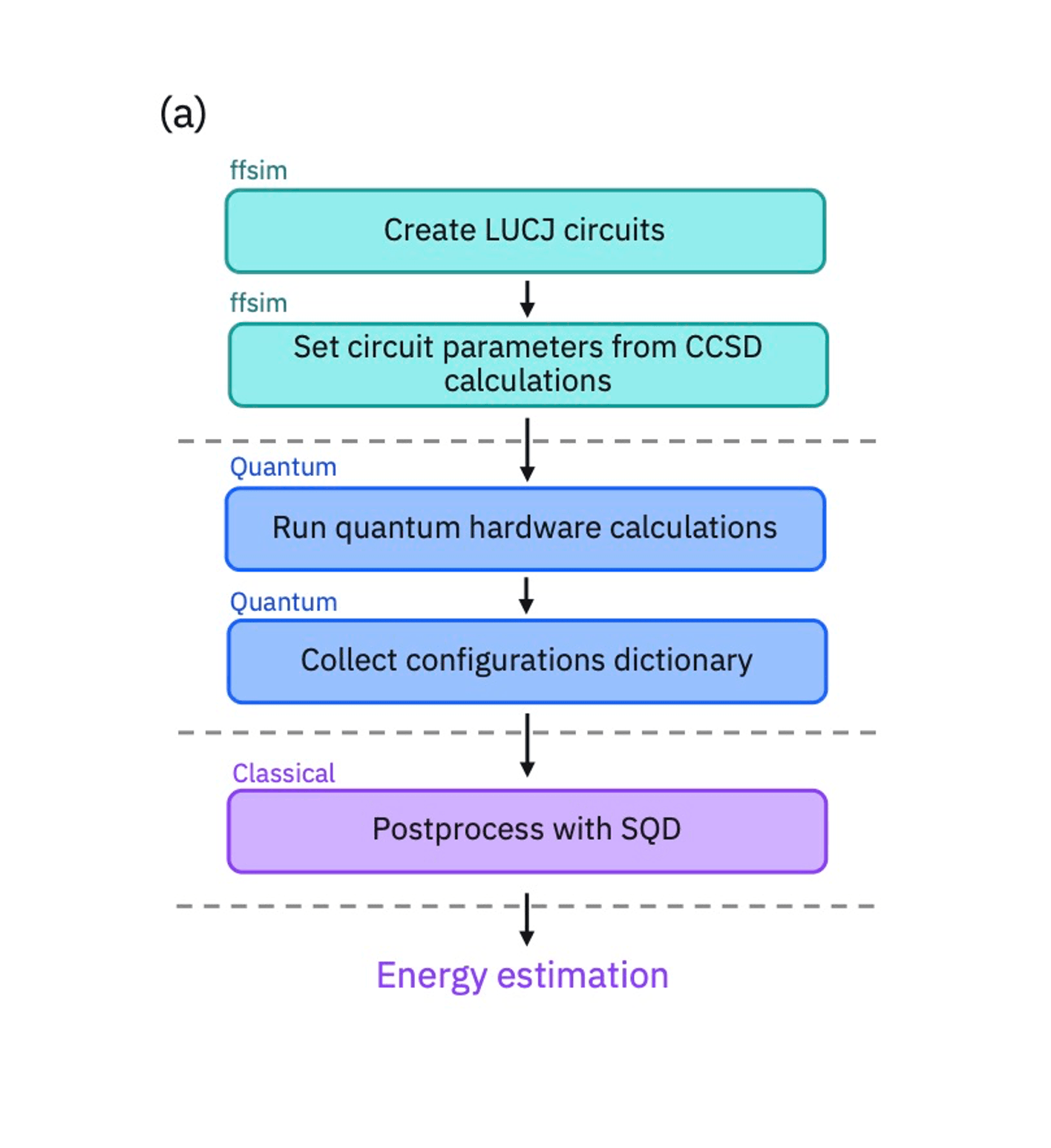
Quantum-centric supercomputing in action
In this study, the team applied the Sample-based Quantum Diagonalization (SQD) method to compute the electronic properties of methylene’s singlet and triplet states. The calculations were performed using 52 qubits of an IBM quantum processor, executing up to 3,000 two-qubit gates per experiment.
This experiment was executed within IBM’s quantum-centric supercomputing framework — a hybrid architecture that tightly couples quantum processors with classical compute resources to enable scalable simulations of molecular systems.
Why it matters
Radical molecules are key players in aerospace, combustion chemistry, and sensor design. Modeling their behavior accurately can lead to better predictive models, more efficient chemical engines, and new sensing technologies capable of detecting minute traces of reactive species.
This study shows that quantum computers are starting to deliver value in real chemical simulations — not just toy problems or idealized systems. As quantum hardware continues to improve and methods like SQD mature, we’re opening the door to modeling complex reaction dynamics and designing better materials with the help of quantum tools.


