.png)

- Article
- Open access
- Published: 11 June 2025
Nature volume 642, pages 356–360 (2025)Cite this article
-
13k Accesses
-
127 Altmetric
Abstract
Compounds consisting only of the element nitrogen (polynitrogens or nitrogen allotropes) are considered promising clean energy-storage materials owing to their immense energy content that is much higher than hydrogen, ammonia or hydrazine, which are in common use, and because they release only harmless nitrogen on decomposition1. However, their extreme instability poses a substantial synthetic challenge and no neutral molecular nitrogen allotrope beyond N2 has been isolated2,3. Here we present the room-temperature preparation of molecular N6 (hexanitrogen) through the gas-phase reaction of chlorine or bromine with silver azide, followed by trapping in argon matrices at 10 K. We also prepared neat N6 as a film at liquid nitrogen temperature (77 K), further indicating its stability. Infrared and ultraviolet–visible (UV-Vis) spectroscopy, 15N-isotope labelling and ab initio computations firmly support our findings. The preparation of a metastable molecular nitrogen allotrope beyond N2 contributes to our fundamental scientific knowledge and possibly opens new opportunities for future energy-storage concepts.
Similar content being viewed by others
Main
Molecular nitrogen allotropes beyond N2 are promising for the development of high-energy-density materials4 because they release enormous energy on dissociation into gaseous N2. As the main component of air, N2 is inert, non-toxic and not a greenhouse contributor5,6,7. Unlike carbon, N2 is the only nitrogen allotrope found in nature and strategies for synthesizing higher neutral molecular nitrogen allotropes are highly sought after8,9,10,11,12,13,14. However, they are deemed extremely unstable, especially when uncharged and with an even electron count15. Consequently, only two examples have been reported. The azide radical (•N3) (Fig. 1a) was identified in the gas phase through rotational spectroscopy in 1956 (refs. 16,17). In 2002, N4 was detected by gas-phase neutralization-reionization mass spectrometry (NRMS); its structure has not been revealed18. The intermediacy of an N6 species was tentatively suggested in 1970 in the decay of azide radicals in aqueous solution but no definitive spectroscopic evidence was provided19.
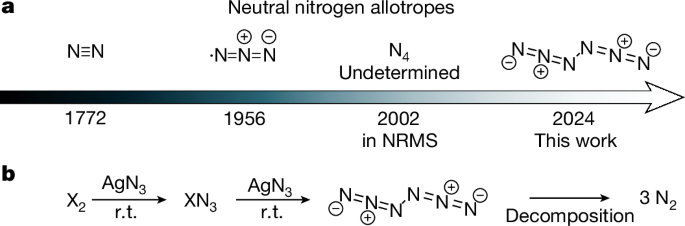
a, Discovery timeline (year given), composition and structure (the structure of N4 has not been determined). b, Reaction sequence used in this study. r.t., room temperature.
There are many computations proposing molecular allotropes spanning from N4 to N120, including chains, rings and cages7,20, most of which have low dissociation barriers into N2. For example, hexazine (cyc-N6, the nitrogen analogue of benzene) exhibits a computed barrier of only 4.2 kcal mol−1 for decomposition into three N2 (ref. 21). Quantum mechanical tunnelling (QMT) effects could further reduce the lifetime of higher nitrogen allotropes, adding to their difficulty of preparation22.
Although the pursuit of higher neutral molecular nitrogen allotropes is extremely challenging, several homonuclear polynitrogen ions have been isolated. The synthesis and characterization of [N5]+[PnF6]− (Pn = As, Sb) salts with a bent pentanitrogen cation represents a milestone23,24. Christe et al. initially identified the \(cyclo{{\rm{-N}}}_{5}^{-}\) anion using mass spectrometry in 2002 and 2003 (refs. 25,26) and Zheng et al. reported in 2017 the synthesis of a salt featuring the \(cyclo{{\rm{-N}}}_{5}^{-}\) anion27. The synthesis of various metal pentazolates was achieved through the reaction of [Na(H2O)(N5)]•2H2O with metal salts28,29.
In the realm of solid-state (non-molecular) structures, a breakthrough was the high-temperature (2,000 K), high-pressure (110 GPa) diamond-like solid-state cubic gauche nitrogen phase in which all atoms are connected by single bonds30,31. An aromatic cyclic hexazine \({{\rm{N}}}_{6}^{4-}\) was identified through solid-state X-ray diffraction of K9N56 under pressures above 40 GPa and temperatures above 2,000 K (ref. 32). Greschner et al. predicted a new nitrogen molecular crystal comprising N6 units with an open-chain structure stabilized by electrostatic interactions33, in line with assessments for the molecular species9.
In our analysis of the proposed molecular nitrogen allotropes, acyclic neutral N6 (hexaaza-1,2,4,5-tetraene, hexanitrogen, diazide) stands out because N2 moieties are not discernible (Fig. 1). The central N–N bond would lead to unproductive endothermic dissociation (ΔG298K theor. +26.1 kcal mol−1, vide infra) into two •N3. Furthermore, the computed dissociation barrier into three N2 molecules of ΔG‡298K = 14.8 kcal mol−1 makes N6 a promising candidate for synthesis. Here we show that N6 indeed can be prepared at room temperature through the reaction of Cl2 or Br2 with AgN3 under reduced pressure, followed by cryogenic trapping34. The characterization was accomplished by infrared (including 15N-isotope labelling) as well as UV-Vis spectroscopy and ab initio computations. We also demonstrate the preparation and stability of C2h-symmetric N6 (hereinafter referred to as N6) in neat form as a film at the temperature of liquid nitrogen (77 K).
Synthesis of N6
As AgN3 is an excellent reagent for the synthesis of polyazides35 and halogen azides both in the gas phase36 and in solution37,38, we suggest that the reaction of AgN3 with XN3 (X = halogen) is a viable route to N6 (Fig. 1b). The reactions were conducted in either a quartz tube or a U-trap by flowing gaseous Cl2 through solid AgN3 under reduced pressure at room temperature (see the ‘Synthesis details’ section in Methods and Supplementary Fig. 1). Apart from the known bands of ClN3 (ref. 39) and HN3 (ref. 40), a distinct group of bands at 2,076.6, 2,049.0, 1,177.6 and 642.1 cm−1 was recorded (Supplementary Fig. 2). After irradiating the matrices with 436 nm light (Fig. 2a middle trace and Supplementary Fig. 3), all bands vanish. However, the rates of decomposition of the newly observed infrared bands differ from those attributed to ClN3 (Supplementary Figs. 4 and 5). There were no discernible products other than chloronitrene (ClN) detected in the difference spectrum after irradiation. Furthermore, identical bands were detected when Br2 was used instead of Cl2, indicating that the unidentified species does not contain halogens (Fig. 2a upper trace and Supplementary Fig. 6). Also, BrN3 does not decompose on 436 nm irradiation, providing clean decomposition spectra of the yet unidentified species.
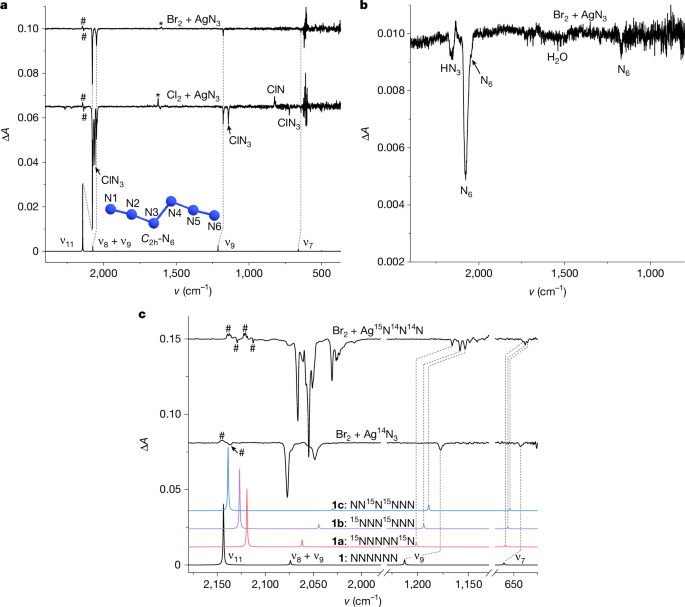
a, Lower trace: computed anharmonic infrared spectrum of N6 at B3LYP/def2-TZVP, including the ν8 + ν9 combination. Middle trace: difference spectrum showing the changes after 8 min of 436 nm irradiation of the products of the reaction of Cl2 with AgN3. Upper trace: difference spectrum showing the changes after 6 min of 436 nm irradiation of the reaction products of Br2 with AgN3. b, Difference spectrum of a neat N6 film at 77 K showing the changes after 8 min of 436 nm irradiation. c, Bottom to top traces: computed anharmonic infrared spectrum of N6, 15NNNNN15N (1a), 15NNN15NNN (1b) and NN15N15NNN (1c) at B3LYP/def2-TZVP, including the ν8 + ν9 combination; difference spectrum showing the changes after 8 min of 436 nm irradiation of the reaction products of Br2 with AgN3; difference spectrum showing changes after 8 min of 436 nm irradiation of the reaction products of Br2 with Ag15N14N14N. Matrix sites from natural abundance and isotope-labelled HN3 (#) and H2O (*) are marked.
The intensive vibrational band at 2,076.6 cm−1 compares favourably with the asymmetric stretching band of the azide moiety in isoelectronic N3–NCO (2,099.1 cm−1, Ar matrix)41. Compared with the computed harmonic vibrations at CCSD(T)/cc-pVTZ, the four bands noted above could be attributed to N6, except the band at 2,049.0 cm−1 of moderate intensity, although it disappeared together with the other bands following photolysis (Supplementary Figs. 4 and 7). To determine the origin of the band at 2,049.0 cm−1, anharmonic vibrational frequencies were computed at B3LYP/def2-TZVP (Supplementary Table 1). This analysis indicates that this band derives from a combination of fundamentals ν8 (ag symmetric N3N4 stretching mode) and ν9 (bu asymmetric N3N2N1 stretching mode). The substantial anharmonic intensity contribution (219 km mol−1; Supplementary Table 2) of the fundamental ν11 at 2,143.5 cm−1 and the ν8 + ν9 combination is notably stronger than its fundamentals, suggesting that the combination ν8 + ν9 gains energy through Fermi resonance from the adjacent strong fundamental ν11 (ref. 41).
To confirm our assignments, isotope-labelling experiments were conducted using Ag15N14N14N. Three groups of distinct peaks can be discerned in the infrared spectra (Fig. 2c and Supplementary Fig. 8), indicating the presence of two N3 moieties in the molecule, which can be attributed to three types of isotopomer (1a: 15NNNNN15N, 1b: 15NNN15NNN, 1c: NN15N15NNN), respectively. In particular, the unsymmetric isotopic substitutions in 1b lower its point group from C2h to Cs. Computations delineate that the terminal (N1 or N6) and internal (N3 or N4) 15N substitutions mainly influence the terminal (ν11) and internal asymmetric stretching vibration (ν9) of the N3 moieties, respectively. This leads to a redshift of the ν8 + ν9 combination and a blueshift of the ν11 fundamental in going from 1a to 1c, resulting in their gradual separation. The intensity ratio of the ν8 + ν9 combination and the ν11 fundamental in 1a is nearly 1:1, which is much higher than that in 1c (about 1:17). These findings align well with the anharmonic infrared intensities computed by density functional theory (Supplementary Table 3), which are attributed to the closer proximity of the ν8 + ν9 combination to the strong ν11 fundamental in 1a, resulting in an increase of the Fermi resonance and vice versa in 1c. Statistically, the anticipated ratio of the three isotopomers should be 1a:1b:1c = 1:2:1, which is reflected in the observed fundamental ν7 in the experimental spectrum (Fig. 3). Furthermore, the computed intensity of ν9 in 1c (107 km mol−1; Supplementary Table 3) is higher than that in 1a (92 km mol−1) and 1b (98 km mol−1), which matches the intensity ratios of ν9 observed in 1a and 1b (approximately 1:2). The experimentally observed intensities agree with these findings and show a slightly higher intensity of ν9 in 1c than in 1a.
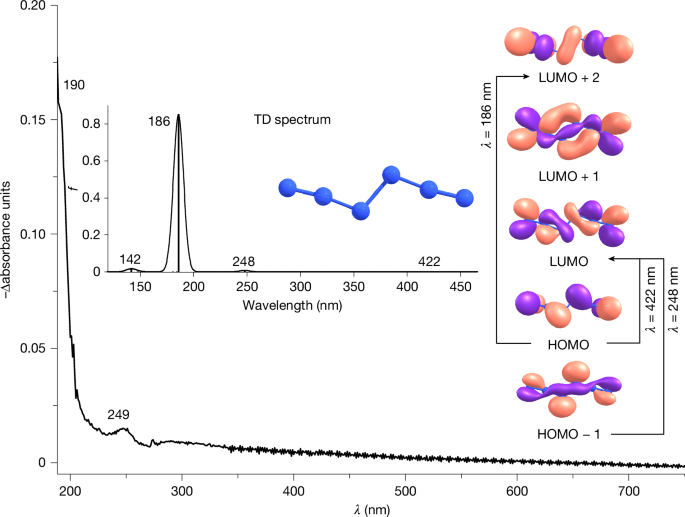
Experimental difference UV-Vis spectrum reflecting changes following 4 min of 436 nm irradiation of the reaction products of Br2 with AgN3 in argon at 10 K. Inset, computed [TD-B3LYP/def2-TZVP] electronic transitions for N6 and molecular orbitals involved.
To explore the intrinsic stability of N6, we also prepared neat N6 at room temperature and condensed it at liquid nitrogen temperature (77 K) as a film on the surface of the matrix window without using argon as a host gas. Irradiation of such N6 films resulted in very similar spectral changes as those observed in argon matrices at 10 K (Fig. 2b and Supplementary Fig. 9). That is, neat N6 is sufficiently stable at the temperature of liquid nitrogen to allow its direct identification.
Further evidence is provided by the UV-Vis spectrum of N6. After 6 min of 436 nm irradiation of the reaction products of Br2 with AgN3, we observed the disappearance of the transitions at 190 and 249 nm and, consistent with the infrared experiments, no new transitions appeared (Fig. 3). All transitions correlate well with the values for the electronic excitations of N6 at 186 nm (f = 0.8512) and 248 nm (f = 0.0078) computed at [TD-B3LYP/def2-TZVP]. Furthermore, the computations reveal a weak electronic excitation at 422 nm (f = 0.0004), corresponding to a π → π* transition, which aligns well with the observed photochemistry.
Computations
To better understand the structure and the potential energy landscape of N6, we computed its energy profile at CCSD(T)/cc-pVTZ (Fig. 4a (ΔG298K) and Supplementary Fig. 10 (ΔH0); see the ‘Computational details’ section in Methods). Only the C2h-N6 trans-conformer is a local minimum; the C2v-N6 cis-conformer is a higher-order stationary point and chemically not relevant42,43. The formal double bond lengths in the N3 moieties are much longer than the triple bond in N2 (theor. 1.104 Å; expt. 1.098 Å)44, indicating double-bond character. Indeed, the computed N2 = N3/N4 = N5 bond length (1.251 Å) is close to that of trans-diazene (HN = NH, theor. 1.253 Å; expt. 1.252 Å)45. The structure of N6 is different from the azide radical (•N = N = N, theor. 1.183 Å; expt. 1.181 Å)46 but comparable with the N3 moiety in hydrazoic acid (HN3, theor. 1.247 and 1.136 Å; expt. 1.237 and 1.133 Å for the N1 = N2 and N2 = N3 bonds, respectively). The N3–N4 bond in N6 (1.460 Å) compares favourably with that in hydrazine (H2N–NH2, theor. 1.445 Å; expt. 1.446 Å). This geometric analysis is well captured by the Lewis structure of N6 (Fig. 1). These conclusions are supported by natural bond orbital computations, which indicate that the terminal nitrogen atoms are electronically neutral, whereas small positive and negative charges are located at N2 and N5 (+0.2e) as well as on N3 and N4 (−0.2e), respectively (Fig. 4a). Equally, N1–N2/N5-N6 have the highest bond order (2.1), followed by N2–N3/N4–N5 (1.4) and N3–N4 (1.1).
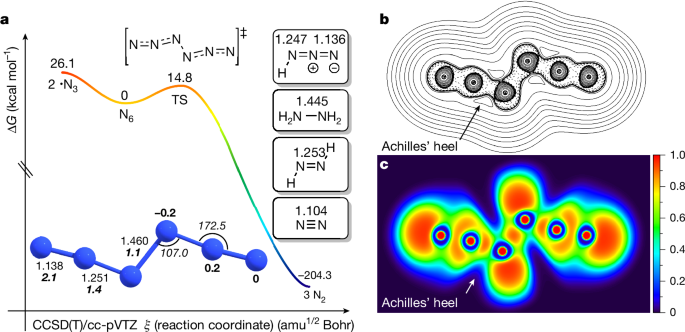
a, Potential energy profile (ΔG298K, kcal mol−1) for N6 at CCSD(T)/cc-pVTZ. The optimized parameters of N6 are given in Ångstrom (normal font), degrees (italics), natural charges in bold and natural bond orders in bold italics. Insets, computed NN bond lengths for N2, trans-HNNH, hydrazine and HN3 at CCSD(T)/cc-pVTZ. b, Contour line map of the Laplacian of the electron density of N6; solid and dashed lines represent positive and negative regions, respectively. c, ELF map.
We visualized the Laplacian of the electron density to gauge where the bonds in N6 are likely to break (Fig. 4b) and why the computed barrier for decomposition into three moles of N2 is, compared with other systems, rather high (ΔG‡298K = 14.8 kcal mol−1). This barrier implies appreciable kinetic stability that is mirrored by our observations. For comparison, the computed barrier of hypothetical D2h-N4 dissociating into two N2 is 6.5 kcal mol−1 at MR-AQCC/VTZ47. With the electron density analysis, the ‘Achilles’ heel’ was discerned at the N2–N3/N4–N5 bonds, as evident from the vertex of positive Laplacian of the in-plane electron density. This is confirmed by the electron localization function (ELF) analysis48 (Fig. 4c). Both the Laplacian of the electron density and the ELF analysis indicate the electron density minimum around the N2–N3/N4–N5 bonds. Hence, even though the Lewis structure would indicate N6 breaking into two •N3 radicals, that is, breaking of the central N3–N4 single bond, the computed barrier for this process amounts to sizeable ΔG298K = 26.1 kcal mol−1 and is unproductive.
On the other hand, ΔG‡298K for the elementary decomposition into three N2 is 14.8 kcal mol−1, implying a finite lifetime of N6 at room temperature. As N6 decomposition may be accelerated by QMT21,22,49, we used canonical variational theory and small-curvature tunnelling computations at B3LYP/def2-TZVP that reveal that N6, unlike hexazine (cyc-N6)21, is unlikely to decompose through QMT, with an estimated half-life of N6 of more than 132 years at 77 K (Supplementary Table 4). At 298 K, the computed half-life still amounts to 35.7 ms. This supports our finding that N6 exists long enough in the gas phase at ambient temperature to be trapped subsequently in cryogenic matrices.
According to CCSD(T)/cc-pVTZ (ΔH0) computations, the decomposition of N6 into three N2 is exothermic (ΔH0) by 185.2 kcal mol−1, which is 2.2 and 1.9 times higher than the decomposition enthalpies of TNT (2,4,6-trinitrotoluene) and HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocane, octogen) by weight50 (see the ‘Computational details’ section in Methods).
We report here the facile synthesis and spectroscopic identification of experimentally unreported hexanitrogen N6. This represents the first, to our knowledge, experimentally realized neutral molecular nitrogen allotrope beyond N2 that exhibits unexpected stability. This discovery challenges the long-held belief of the elusiveness of neutral molecular nitrogen allotropes.
Methods
Matrix apparatus design
For the matrix isolation studies, we used an APD Cryogenics HC-2 cryostat with a closed-cycle refrigerator system, equipped with an inner CsI window for infrared measurements. Spectra were recorded at the temperature of the matrix (10 K) with a Bruker VERTEX 70 FT-IR spectrometer with a spectral range of 4,000–400 cm−1 and a resolution of 0.7 cm−1 and UV-Vis spectra were recorded with a Jasco V-670 spectrophotometer equipped with an inner sapphire window. A high-pressure mercury lamp (HBO 200, Osram) with a monochromator (Bausch & Lomb) was used for irradiation. Cl2 or Br2 was evaporated (Cl2-CCl4: −140 °C, Br2: −85 °C) from a storage bulb into the quartz tube or U-trap. Although not directly measured, all reaction products were co-condensed with a large excess of argon (typically 60–120 mbar from a 2,000-ml storage bulb) onto the surface of the matrix window at 10 K in several milliseconds.
Synthesis details
Warning! Silver azide and halogen azides are extremely hazardous and explosive. Such compounds should be handled with utmost care and only in very small quantities (<5 mmol). Appropriate safety precautions (blast screens, face shields, Kevlar gloves, soundproof earmuffs and protective leather clothing) are necessary. Make sure to eliminate static electricity before handling. It is also crucial to avoid friction and light exposure and prevent any contact with metals during sample handling to ensure safety.
Silver azide was synthesized by adding a stoichiometric amount of a silver nitrate–water solution to a sodium azide–water solution in the dark. The precipitate was washed three times with anhydrous ethanol. The resulting slurry was loosely dispersed on one side of the inner surface of a straight quartz tube (ø 10 × 1) or the inner surface of a U-trap (inside diameter 10 mm) and then brought to reduced pressure to remove the solvent. Typically, 0.6 mmol and 2.5 mmol of AgN3 are required for the straight quartz tube and U-trap, respectively. Na15N14N14N (>99% 15N, Sigma-Aldridge) was used for isotope labelling experiments. Chlorine gas was bubbled into CCl4 at 0 °C and degassed before use. Bromine was purified by vacuum distillation before use. Typically, 3 mmol of halogen were stored in the storage bulb for the reaction.
Computational details
Geometry optimizations and energy computations were carried out at the CCSD(T)/cc-pVTZ51,52,53 levels of theory using ORCA 5.0 (with keywords verytightscf and verytightopt)54. B3LYP55,56 computations (geometry optimizations, energy computations (all free energies were computed at 298 K), harmonic vibrational analysis and DVPT2 anharmonic vibrational analysis) were performed using Gaussian 16 (ref. 57) with a def2-TZVP basis set58. Local minima were confirmed by vibrational frequencies analyses and transition states were further confirmed by intrinsic reaction coordinate computations. Harmonic vibrational analysis at CCSD(T)/cc-pVTZ was performed using CFOUR v2.1 (ref. 59). Wavefunction analysis (Laplacian of electron density and electron localization function) results were obtained from Multiwfn 3.8 (ref. 60) at CCSD(T)/cc-pVTZ. Natural bond order analysis and resonance structures were computed with NBO 7.0 (refs. 61,62). CVT/SCT (canonical variational transition state theory with small-curvature tunnelling) and CVT/ZCT (canonical variational transition state theory with zero-curvature tunnelling) computations were carried out with Gaussrate 17 (refs. 27,63,64,65,66,67) as an interface between Gaussian 16 and Polyrate68. Furthermore, local stretching force constants were obtained by LModeA-nano69 as a plugin of the open-source version of the visualization program PyMOL.
Detonation calculation details
First, the density (ρ, in cm3 per molecule) of the N6 crystal was determined using electrostatic interaction correction as suggested by Politzer et al.70 (equation (2)). Mm (84.04/(6.02 × 1023) g per molecule) is the molecular mass. Vm (610.52/(1.89 × 108)3 cm3 per molecule) is the volume of the isolated gas-phase molecule, which was determined by the 0.001 a.u. density envelope using the marching tetrahedron method60,71. ν is the parameter of balance between positive and negative surface potentials72 (equation (2)). \({\sigma }_{{\rm{tot}}}^{2}\) (48.40 kcal2 mol−2) is the strengths and variabilities of the overall surface potentials, which could be derived from variance of positive (\({\sigma }_{+}^{2}\), 31.06 kcal2 mol−2) and negative charges (\({\sigma }_{-}^{2}\), 17.34 kcal2 mol−2) with equation (3). α (0.9183), β (0.0028) and γ (0.0443) are coefficients.
$$\rho =\alpha \left(\frac{{M}_{{\rm{m}}}}{{V}_{{\rm{m}}}}\right)+\beta (\nu {\sigma }_{{\rm{tot}}}^{2})+\gamma $$
(1)
$$\nu =\frac{{\sigma }_{+}^{2}{\sigma }_{-}^{2}}{{({\sigma }_{+}^{2}+{\sigma }_{-}^{2})}^{2}}$$
(2)
$${\sigma }_{{\rm{tot}}}^{2}={\sigma }_{+}^{2}+{\sigma }_{-}^{2}$$
(3)
The detonation velocity (D) and detonation pressure (P) were calculated using the Kamlet–Jacobs equation73 (equations (4) and (5)). N is the number of moles of the gas generated per gram (equation (6)), \(\bar{M}\) is the average molecular weight of the gaseous product (equation (7)), Q is the heat of detonation (equation (8)), M is the molecular weight (84.04 g mol−1), ΔHf is the standard heat of formation (774.88 kJ mol−1, which was derived from the energy difference of the computed enthalpy at 298 K between C2h-N6 and 3 moles of N2) and a (0), b (0), c (0) and d (6) represent the number of C, H, O, and N atoms in the molecule, respectively.
$$D=1.01{(N\sqrt{\bar{M}Q})}^{\frac{1}{2}}(1+1.3\rho )$$
(4)
$$P=1.558{\rho }^{2}N\sqrt{\bar{M}Q}$$
(5)
$$N=\frac{b+2c+2d}{4M}$$
(6)
$$\bar{M}=\frac{4M}{b+2c+2d}$$
(7)
$$Q=\frac{28.9b+94.05a+0.239\Delta {H}_{{\rm{f}}}}{M}$$
(8)
Assessing the energetic performance using the Kamlet–Jacobs equation73, the CCSD(T)/cc-pVTZ level of theory predicts a lower density (ρ: 1.51 g cm−3) than that of TNT (1.65 g cm−3) and an excellent detonation performance (detonation velocity D: 8,930 m s−1; detonation pressure P: 31.7 GPa). This compares favourably with several well-known explosives, for example, TNT (D: 6,900 m s−1; P: 21.0 GPa), RDX (1,3,5-trinitro-1,3,5-triazinane; D: 8,750 m s−1; P: 34.5 GPa) and FOX-7 (1,1-diamino-2,2-dinitroethylene; D: 8,870 m s−1; P: 34.5 GPa)74.
Energy-releasing equivalent calculation details
A kiloton of N6 is 1.19 × 107 mol, which can release an energy of 2.20 × 109 kcal (9.21 terajoules) based on the enthalpy (ΔH0). Considering that the standard kiloton TNT equivalent is 4.184 terajoules, N6 can release 2.2 times the energy of TNT of the same weight. On the basis of the documented TNT equivalent based on weight for HMX (1.15) and RDX (1.15)50, N6 can release 1.9 times the energy of HMX or RDX with the same weight.
References
Christe, K. O. Polynitrogen chemistry enters the ring. Science 355, 351–351 (2017).
Wang, Y. et al. Stabilization of hexazine rings in potassium polynitride at high pressure. Nat. Chem. 14, 794–800 (2022).
Ninet, S. Benzene-like N6 hexazine rings. Nat. Chem. 15, 595–596 (2023).
Yao, Y. & Adeniyi, A. O. Solid nitrogen and nitrogen‐rich compounds as high‐energy‐density materials. Phys. Status Solidi B 258, 2000588 (2021).
Klapötke, T. M. & Witkowski, T. G. Nitrogen-rich energetic 1,2,5-oxadiazole-tetrazole-based energetic materials. Propellants Explos. Pyrotech. 40, 366–373 (2015).
Nguyen, M. T. Polynitrogen compounds: 1. Structure and stability of N4 and N5 systems. Coord. Chem. Rev. 244, 93–113 (2003).
Zarko, V. E. Searching for ways to create energetic materials based on polynitrogen compounds (review). Combust. Explos. Shock Waves 46, 121–131 (2010).
Larson, Å., Larsson, M. & Östmark, H. Theoretical study of rectangular (D2h) N4. J. Chem. Soc. Faraday Trans. 93, 2963–2966 (1997).
Glukhovtsev, M. N. & von Ragué Schleyer, P. Structures, bonding and energies of N6 isomers. Chem. Phys. Lett. 198, 547–554 (1992).
Glukhovtsev, M. N., Jiao, H. & von Ragué Schleyer, P. Besides N2, what is the most stable molecule composed only of nitrogen atoms? Inorg. Chem. 35, 7124–7133 (1996).
Strout, D. L. Acyclic N10 fails as a high energy density material. J. Phys. Chem. A 106, 816–818 (2002).
Hirshberg, B., Gerber, R. B. & Krylov, A. I. Calculations predict a stable molecular crystal of N8. Nat. Chem. 6, 52–56 (2014).
Strout, D. L. Cage isomers of N14 and N16: nitrogen molecules that are not a multiple of six. J. Phys. Chem. A 108, 10911–10916 (2004).
Samartzis, P. C. & Wodtke, A. M. All-nitrogen chemistry: how far are we from N60? Int. Rev. Phys. Chem. 25, 527–552 (2010).
Mikhailov, O. V. Molecular and electronic structures of neutral polynitrogens: review on the theory and experiment in 21st century. Int. J. Mol. Sci. 23, 2841 (2022).
Thrush, B. A. & Norrish, R. G. W. The detection of free radicals in the high intensity photolysis of hydrogen azide. Proc. R. Soc. Lond. A 235, 143–147 (1956).
Beaman, R. A., Nelson, T., Richards, D. S. & Setser, D. W. Observation of azido radical by laser-induced fluorescence. J. Phys. Chem. 91, 6090–6092 (1987).
Cacace, F., de Petris, G. & Troiani, A. Experimental detection of tetranitrogen. Science 295, 480–481 (2002).
Hayon, E. & Simic, M. Absorption spectra and kinetics of the intermediate produced from the decay of azide radicals. J. Am. Chem. Soc. 92, 7486–7487 (1970).
Zhou, H., Wong, N.-B., Zhou, G. & Tian, A. Theoretical study on “multilayer” nitrogen cages. J. Phys. Chem. A 110, 3845–3852 (2006).
Sedgi, I. & Kozuch, S. Quantum tunneling instability of the mythical hexazine and pentazine. Chem. Commun. 60, 2038–2041 (2024).
Schreiner, P. R. Quantum mechanical tunneling is essential to understanding chemical reactivity. Trends Chem. 2, 980–989 (2020).
Christe, K. O., Wilson, W. W., Sheehy, J. A. & Boatz, J. A. N5+: a novel homoleptic polynitrogen ion as a high energy density material. Angew. Chem. Int. Ed. 38, 2004–2009 (1999).
Vij, A. et al. Polynitrogen chemistry. Synthesis, characterization, and crystal structure of surprisingly stable fluoroantimonate salts of N5+. J. Am. Chem. Soc. 123, 6308–6313 (2001).
Vij, A., Pavlovich, J. G., Wilson, W. W., Vij, V. & Christe, K. O. Experimental detection of the pentaazacyclopentadienide (pentazolate) anion, cyclo-N5−. Angew. Chem. Int. Ed. 41, 3051–3054 (2002).
Östmark, H. et al. Detection of pentazolate anion (cyclo-N5−) from laser ionization and decomposition of solid p-dimethylaminophenylpentazole. Chem. Phys. Lett. 379, 539–546 (2003).
Zhang, C. et al. Synthesis and characterization of the pentazolate anion cyclo-N5ˉ in (N5)6(H3O)3(NH4)4Cl. Science 355, 374–376 (2017).
Xu, Y. et al. A series of energetic metal pentazolate hydrates. Nature 549, 78–81 (2017).
Xu, Y., Tian, L., Li, D., Wang, P. & Lu, M. A series of energetic cyclo-pentazolate salts: rapid synthesis, characterization, and promising performance. J. Mater. Chem. A 7, 12468–12479 (2019).
Eremets, M. I., Gavriliuk, A. G., Trojan, I. A., Dzivenko, D. A. & Boehler, R. Single-bonded cubic form of nitrogen. Nat. Mater. 3, 558–563 (2004).
Benchafia, E. M. et al. Cubic gauche polymeric nitrogen under ambient conditions. Nat. Commun. 8, 930 (2017).
Laniel, D. et al. Aromatic hexazine [N6]4− anion featured in the complex structure of the high-pressure potassium nitrogen compound K9N56. Nat. Chem. 15, 641–646 (2023).
Greschner, M. J. et al. A new allotrope of nitrogen as high-energy density material. J. Phys. Chem. A 120, 2920–2925 (2016).
Qian, W. Y., Mardyukov, A. & Schreiner, P. R. Hexanitrogen (N6): a synthetic leap towards neutral nitrogen allotropes. Preprint at https://doi.org/10.26434/chemrxiv-2024-90vvx (2024).
Zeng, X. et al. Reaction of AgN3 with SOCl2: evidence for the formation of thionyl azide, SO(N3)2. Inorg. Chem. 43, 4799–4801 (2004).
Raschig, F. Über Chlorazid N3Cl. Ber. Dtsch. Chem. Ges. 41, 4194–4195 (1908).
Lyhs, B., Bläser, D., Wölper, C., Schulz, S. & Jansen, G. A comparison of the solid-state structures of halogen azides XN3 (X=Cl, Br, I). Angew. Chem. Int. Ed. 51, 12859–12863 (2012).
Buzek, P., Klapötke, T. M., von Ragué Schleyer, P., Tornieporth‐Oetting, I. C. & White, P. S. Iodine azide. Angew. Chem. Int. Ed. 32, 275–277 (1993).
Shurvell, H. F. & Hyslop, D. W. Infrared spectrum of cyanogen azide. J. Chem. Phys. 52, 881–887 (1970).
Pimental, G. C. & Charles, S. W. Infrared spectral perturbations in matrix experiments. Pure Appl. Chem. 7, 111–124 (1963).
Zeng, X., Beckers, H. & Willner, H. Matrix isolation of two isomers of N4CO. Angew. Chem. Int. Ed. Engl. 50, 482–485 (2011).
Tobita, M. & Bartlett, R. J. Structure and stability of N6 isomers and their spectroscopic characteristics. J. Phys. Chem. A 105, 4107–4113 (2001).
Gagliardi, L., Evangelisti, S., Barone, V. & Roos, B. O. On the dissociation of N6 into 3 N2 molecules. Chem. Phys. Lett. 320, 518–522 (2000).
Huber, K. P. & Herzberg, G. in Molecular Spectra and Molecular Structure (eds Huber, K. P. & Herzberg, G.) Ch. 2, 8–689 (Springer, 1979).
Carlotti, M., Johns, J. W. C. & Trombetti, A. The ν5 fundamental bands of N2H2 and N2D2. Can. J. Phys. 52, 340–344 (1974).
Brazier, C. R., Bernath, P. F., Burkholder, J. B. & Howard, C. J. Fourier transform spectroscopy of the ν3 band of the N3 radical. J. Chem. Phys. 89, 1762–1767 (1988).
Bittererová, M., Östmark, H. & Brinck, T. Ab initio study of the ground state and the first excited state of the rectangular (D2h)N4 molecule. Chem. Phys. Lett. 347, 220–228 (2001).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Schreiner, P. R. Tunneling control of chemical reactions: the third reactivity paradigm. J. Am. Chem. Soc. 139, 15276–15283 (2017).
Weggel, D. C. in Blast Protection of Civil Infrastructures and Vehicles Using Composites (ed. Uddin, N.) 3–43 (Woodhead Publishing, 2010).
Pople, J. A., Head‐Gordon, M. & Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 87, 5968–5975 (1987).
Bartlett, R. J. & Purvis, G. D. Many-body perturbation theory, coupled-pair many-electron theory, and the importance of quadruple excitations for the correlation problem. Int. J. Mol. Sci. 14, 561–581 (1978).
Pople, J. A., Krishnan, R., Schlegel, H. B. & Binkley, J. S. Electron correlation theories and their application to the study of simple reaction potential surfaces. Int. J. Mol. Sci. 14, 545–560 (1978).
Neese, F. Software update: the ORCA program system—version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Becke, A. D. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. & Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994).
Frisch, M. J. et al. Gaussian 16, Revision B.01 (Gaussian, Inc., 2016).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Stanton, J. F. et al. CFOUR, coupled-cluster techniques for computational chemistry, a quantum-chemical program package with the integral packages MOLECULE (J. Almlöf and PR Taylor), PROPS (PR Taylor) (2014).
Lu, T. & Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 38, 314–323 (2012).
Glendening, E. D., Landis, C. R. & Weinhold, F. NBO 7.0: new vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 40, 2234–2241 (2019).
Glendening, E. D., Landis, C. R. & Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2, 1–42 (2012).
Zheng, J. et al. Gaussrate 17-B (Univ. Minnesota, 2017).
Garrett, B. C. & Truhlar, D. G. Generalized transition state theory. Bond energy-bond order method for canonical variational calculations with application to hydrogen atom transfer reactions. J. Am. Chem. Soc. 101, 4534–4548 (1979).
Garrett, B. C. & Truhlar, D. G. Criterion of minimum state density in the transition state theory of bimolecular reactions. J. Chem. Phys. 70, 1593–1598 (1979).
Garrett, B. C., Truhlar, D. G., Grev, R. S. & Magnuson, A. W. Improved treatment of threshold contributions in variational transition-state theory. J. Phys. Chem. 84, 1730–1748 (1980).
Truhlar, D. G., Issacson, A., Skodje, R. & Garrett, B. C. Additions and corrections - incorporation of quantum effects in generalized-transition-state theory. J. Phys. Chem. 87, 4554–4554 (1983).
Zheng, J. et al. Polyrate-version 2017-C (Univ. Minnesota, 2017).
Tao, Y., Zou, W., Nanayakkara, S. & Kraka, E. LModeA-nano: a PyMOL plugin for calculating bond strength in solids, surfaces, and molecules via local vibrational mode analysis. J. Chem. Theory Comput. 18, 1821–1837 (2022).
Politzer, P., Martinez, J., Murray, J. S., Concha, M. C. & Toro-Labbé, A. An electrostatic interaction correction for improved crystal density prediction. Mol. Phys. 107, 2095–2101 (2009).
Bader, R. F. W., Carroll, M. T., Cheeseman, J. R. & Chang, C. Properties of atoms in molecules: atomic volumes. J. Am. Chem. Soc. 109, 7968–7979 (1987).
Murray, J. S., Concha, M. C. & Politzer, P. Links between surface electrostatic potentials of energetic molecules, impact sensitivities and C–NO2/N–NO2 bond dissociation energies. Mol. Phys. 107, 89–97 (2009).
Kamlet, M. J. & Jacobs, S. J. Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O explosives. J. Chem. Phys. 48, 23–35 (1968).
Prazyan, T. L. & Zhuravlev, Y. N. Computer simulation of the structure and electronic and detonation properties of energy materials. Combust. Explos. Shock Waves 53, 718–723 (2017).
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG) through grant MA 8773/3-1 (A.M.) is gratefully acknowledged. We sincerely thank X. Bi (Northeast Normal University), T. M. Klapötke (Ludwig Maximilian University of Munich) and Y. Ning (Northeast Normal University) for their valuable suggestions for the synthesis of silver azide and T. Lu (Beijing Kein Research Center for Natural Sciences) for advice about the detonation performance computations. We also thank W. D. Allen (University of Georgia) and X. Zeng (Fudan University) for carefully reading and commenting on the manuscript.
Funding
Open access funding provided by Justus-Liebig-Universität Gießen.
Ethics declarations
Competing interests
W.Q., A.M., and P.R.S. are inventors on European patent application EP24194869 (16 August 2024), submitted by the Justus Liebig University Giessen, which covers a method for producing molecular polynitrogens.
Peer review
Peer review information
Nature thanks Chunlin He, Thomas Klapötke and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains a note for the synthesis, Supplementary Figs. 1–15, Supplementary Tables 1–4 as well as xyz-coordinates of all computed species.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Qian, W., Mardyukov, A. & Schreiner, P.R. Preparation of a neutral nitrogen allotrope hexanitrogen C2h-N6 . Nature 642, 356–360 (2025). https://doi.org/10.1038/s41586-025-09032-9
Received: 18 September 2024
Accepted: 16 April 2025
Published: 11 June 2025
Issue Date: 12 June 2025
DOI: https://doi.org/10.1038/s41586-025-09032-9


