.png)


There is a very famous essay titled ‘Reality has a surprising amount of detail’. The thesis of the article is that reality is filled, just filled, with an incomprehensible amount of materially important information, far more than most people would naively expect. Some of this detail is inherent in the physical structure of the universe, and the rest of it has been generated by centuries of passionate humans imbibing the subject with idiosyncratic convention. In either case, the detail is very, very important. A wooden table is “just” a flat slab of wood on legs until you try building one at industrial scales, and then you realize that a flat slab of wood on legs is but one consideration amongst grain, joint stability, humidity effects, varnishes, fastener types, ergonomics, and design aesthetics. And this is the case for literally everything in the universe.
Including cancer.
But up until just the last few centuries, it wasn’t really treated that way. It was only in the mid-1800’s when Rudolf Virchow, the father of modern pathology, realized that despite most forms of cancer looking reasonably similar to the naked eye, they were—under the microscope—anything but uniform. There was squamous carcinoma with its jagged islands of keratinizing cells, adenocarcinoma with its glandular tubes, sarcoma with its spindle-shaped whorls. And, as a generation of pathologists began to train, they also noticed that visual appearance of cancer often seemed to correlate with how slow, aggressive, quiet, violent, widespread, or local the disease ended up being. Over time, those clues accumulated into prognostic systems: Broders’ classification for squamous carcinoma, Bloom-Richardson for breast cancer, Gleason for prostate. What began as an intuitive visual ‘feeling’ became codified into scales that pathologists across the world could consistently apply.
Then, it was noticed that even the genetic material contained within tumor cells were aberrant, their misshapen karyotypes so obvious as to be even visible under a light microscope. In 1960, the discovery of the “Philadelphia chromosome” in chronic myeloid leukemia marked the first consistent, disease-defining genetic quirk of cancer: a translocation between chromosomes 9 and 22. This was not the only one. In the decades that passed during the following genetic sequencing revolution, a great deal of oncogenes (if mutated or overexpressed, cause cancer) and tumor suppressor genes (if lost, cause cancer) were identified and catalogued away. Many of them are immediately recognizable by undergraduate biology students: KRAS, p53, RB1, MYC, and so on. Some of them, namely the BRCA set of mutations, have even entered common parlance as synonymous with inherited cancer risk.
The next jump was in the world of proteins. Sometimes cancers heavily altered genetic sequence is enough to tell you something interesting. But, other times, the genetic sequence itself hasn’t changed much, rather, the major alteration is in how little or how high a gene is transcribed to protein. With the advent of immunofluorescence, the study of protein abundance in tissue became possible to study at scale.
During the 1980’s, this study of cancer proteins was used by Axel Ullrich, a scientist at Genentech, and Dennis Slamon, an oncologist at UCLA, to help create one of the most successful oncologic drugs of all time. Axel’s research at Genentech had established HER2 as a particularly aggressive oncogene. At the same time, Dennis’s analyses using UCLA patient records showed that patients whose tumors had HER2 overexpression, determined by protein expression, consistently had worse outcomes. A coincidental discussion between the two led to an obvious question: if HER2 was driving the aggressiveness, could a drug directly target it?
This led to the development of trastuzumab, or Herceptin, an antibody against the HER2 receptor, blocking its activity. In clinical trials led by Slamon in the 1990s, women with HER2-positive tumors, previously consigned to dismal prognoses, now lived far longer when trastuzumab was added to their chemotherapy. Today, trastuzumab is still heavily relied upon, and there are now dozens of other drugs underneath the HER2-targeting umbrella.
But even amongst modern analogues, it still only works in the patients whose tumors bear the detail. If a breast cancer is HER2-negative, trastuzumab is useless, even harmful. If it is HER2-positive, the drug can be life-saving. Consider the following image. The ‘naked’ view of cancer is H&E, or the top row. There, A, B, and C don’t look particularly different, do they?
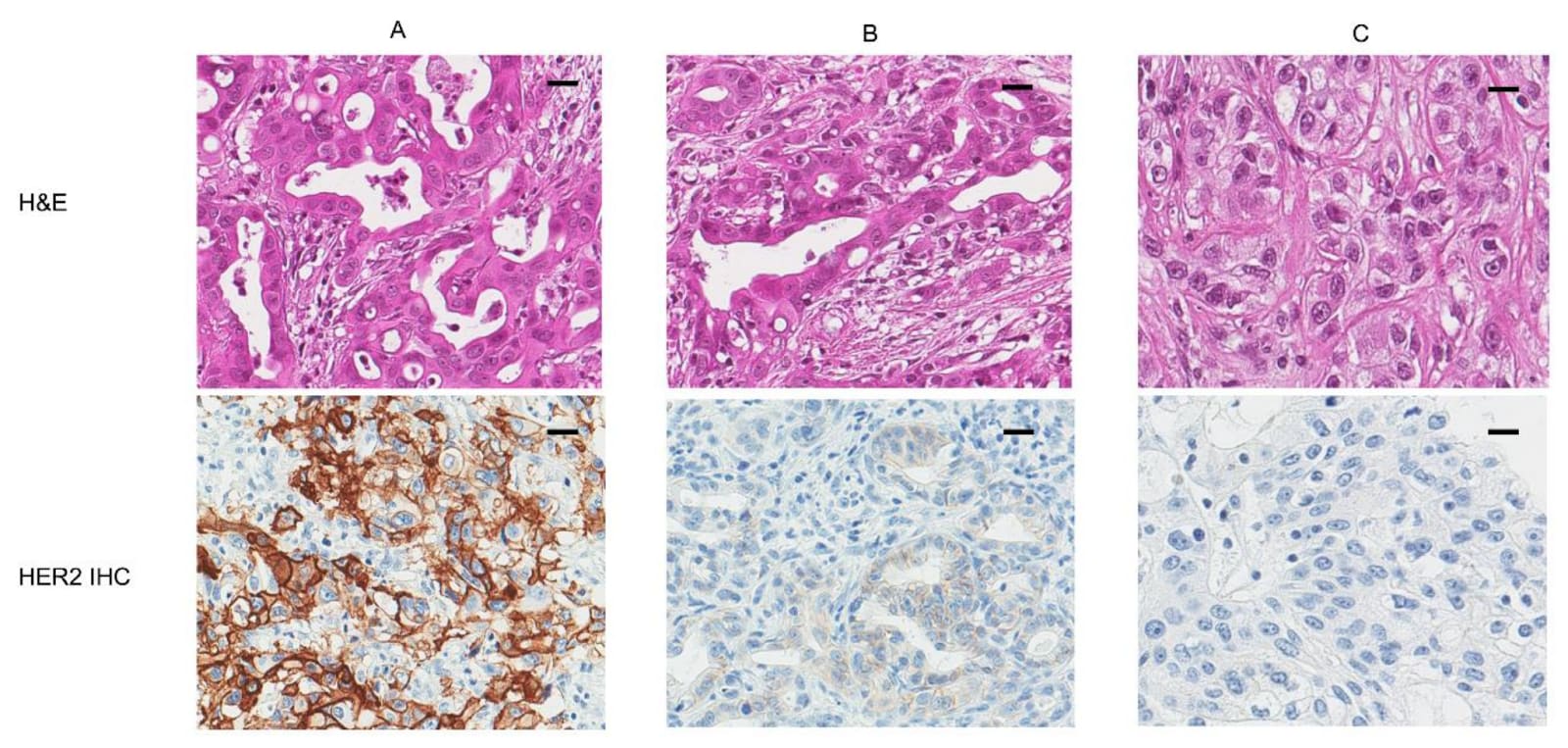
It is only in looking at the bottom row, where the patient’s tumor cells have been stained with an antibody specifically meant to bind to HER2, that a clinician would know that only patient A would respond to the drug, patient B may slightly respond, and patient C wouldn’t respond at all.
It’s worth being quite astonished at this, because there is very little else like it amongst all human maladies. As in, where entire lines of treatment may fail to work due to an extremely subtle biological phenomenon that, up until a few decades back, science wasn’t even aware of, let alone quantify. In most areas of medicine, a diagnosis tends to unify patients under a single therapeutic approach: antibiotics for bacterial pneumonia, insulin for type 1 diabetes, thyroid hormone for hypothyroidism. The drug may differ in dose or formulation, but not in principle.
This is not the case for cancer.
In fact, the story of trastuzumab is not particularly unique, it has repeated again and again and again. Tamoxifen is revolutionary, but only works in ER-positive breast cancer, where the tumor cells are dependent on estrogen signaling. Keytruda is revolutionary, but primarily works in PD-L1 positive cancers, where the tumor microenvironment has upregulated immune checkpoints as a shield against T cells. Tagrisso is revolutionary, but only works in lung cancers where certain genetic mutations are present. And so on.
Cancer, in many ways, is among one of the most detailed diseases on earth.
As of today, much of the ‘cancer understanding’ literature is exactly what has gone on for the past hundred-or-so years, just scaled up. Whereas immunofluorescence allowed you to paint one or two proteins onto a tumor section, multiplexed imaging can now overlay forty. Where HER2 or ER were once binary categories, modern day RNA sequencing can reveal thousands of differentially expressed genes, each with subtle implications. And these methods can be pushed to the spatial dimension too, producing maps of gene/protein expression across entire tumors, showing not just what is “on” or “off,” but exactly where and in what neighborhood.
This is great, because cancer is obviously still a major problem facing humanity. Pancreatic cancer still has a 5-year survival rate of just ~13% and lung cancer has a 5-year survival of 9% if metastasized. And some of that surely comes down to us simply not understanding cancer well enough; consider the fact that about 44% of U.S. cancer patients were nominally eligible for an immune checkpoint inhibitor and an estimated ~12.5% actually benefited.
And so work has gone on to learn more, and learn more we have.
We’re finding that spatial organization of CCR7+ dendritic cells in tumors helps predict pembrolizumab response in head and neck cancer. We’re finding that B-cells being localized within so-called tertiary lymphoid structures seem to improve immune checkpoint blockade efficacy. We’re finding that higher CD34 expression in macrophage-dense regions of a tumor correlates with a worse response to camrelizumab. I think one of the craziest things we’ve found is that tumor cells can pump out exosomes—tiny lipid vesicles—carrying microRNAs that reprogram distant tissues into pre-metastatic niches before a single malignant cell arrives; the existence of which can predict response to a great deal of chemotherapies and immunotherapies.
All this is very exciting work. Unfortunately, basically none of it has been turned into anything clinically useful.
I’m not the first to notice this. In the 2010s, there were a flurry of papers bemoaning this exact phenomenon: The failure of protein cancer biomarkers to reach the clinic, Why your new cancer biomarker may never work, and Waste, leaks, and failures in the biomarker pipeline. The first paper has a particularly illustrating line:
…very few, if any, new circulating cancer biomarkers have entered the clinic in the last 30 years. The vast majority of clinically useful cancer biomarkers were discovered between the mid-1960s (for example, carcinoembryonic antigen, CEA) and the early 1980s (for example, prostate-specific antigen (PSA) and carbohydrate antigen 125 (CA125)).
Though these papers were written a decade-or-so back, I can’t find any evidence that there have been any significant breakthroughs since then, with perhaps the exception of cfDNA, or cell-free DNA, though this is still being proven out.
The blame for this is heterogenous. A lot of the aforementioned papers discuss how newer biomarkers often have shoddy validation, need more datapoints, have variable accuracy, or are so biologically implausible as to likely be an artifact of the underlying data. I don’t disagree with any of these, the replication crisis is as real in the cancer biomarker literature as it is anywhere else. But I’d like to focus on one fault that all the papers mention: the inability for many novel biomarkers to improve on the current clinical standard.
I think it is unlikely that any singular biomarker developed after the 1980s will do this. And we shouldn’t expect it to.
Cancer, like everything else in the universe, is defined by a set of rules, a set of universalities. Biologists love to talk about how biology as a domain is filled with exceptions, but even exceptions themselves are rules. In our effort to understand the disease, we have gathered many rules, some of which have been discussed here: HER2, PD-L1, and the like. The field, likely for decades, hoped that these seemingly simple biomarkers were just the tip of the iceberg, and with enough data, enough pouring over the numbers, we’d stumble across something more fundamental about cancer; the rest of the iceberg.
This has not been the case. Increasingly, it is seeming like these ‘obvious’ biomarkers do, empirically, account for a great deal of what matters in cancer. Unlike physics, cancer never offered much “room at the bottom”—at least not in the sense of yielding endless layers of clinically useful, legible rules.
Phrased differently: if our existing rules explain, say, 60% of the between-patient variance, how is it possible that any new biomarker could swoop in and shoulder the rest on its own? It cannot. It empirically cannot.
But none of this is to say that it is not worth trying to understand the remaining variance, just that it will require a different strategy.
The situation here is not dissimilar to language. Knowing the meaning of a single word tells you something, but not nearly enough to understand a sentence, much less a paragraph. Meaning emerges from combinations, syntax, context, and emphasis. Cancer is the same. “HER2-positive” is a word. “HER2-positive, PD-L1-high, tumor-mutational-burden-high, tertiary-lymphoid-structure present, with exhausted CD8 niches” is a sentence. Words are enough to get you quite far, but if you wish to operate in the long-tails (where we currently are with cancer!), then it is insufficient. The field has spent the last few centuries compiling the words, but now it is time to learn the grammar, the joint-distribution of every word in combination with every other word.
In other words, the obvious next step is to stop asking for singular biomarkers to bear the entire burden of explanation, and instead ask how many small signals can be woven into a coherent, usable picture. But this creates a combinatorial explosion! If you have 20 binary biomarkers, that’s over a million possible patient subgroups. No trial, no matter how well-funded, can enumerate that space.
How can we escape this problem? It is increasingly my opinion that the only reasonable path forward is to delegate the problem of cancer biomarkers to machine intelligence. Rely on the compression, abstraction, and pattern-finding abilities of statistical models that can hold dozens, hundreds, thousand weak signals in memory at once, and then distill them down into single, actionable scores.
This may sound far-fetched, but realistically speaking, it has been going on for some time now. Multigene expression panels from the early 2000s, like OncotypeDX or MammaPrint were, in spirit, primitive machine-learning models: linear combinations of weak features, trained against outcomes, that outperform any single gene.
And in recent years, it is accelerating even further.
For example, you may be aware that the aforementioned BRCA mutations, a massive driver of breast cancer risk, causes homologous recombination deficiency (HRD), or, the inability to faithfully repair double-strand breaks in DNA. In turn, this often causes cancer. But what may be a surprise is that BRCA mutations aren’t the only way that a patient could have HRD, many other genes in the homologous recombination repair pathway—PALB2, RAD51C, RAD51D, FANCA, ATM, CHEK2, and more—can be mutated, leading to the exact same phenotype. Even promoter methylation of BRCA1 (with the gene intact but “turned off”) can produce HRD. And knowing whether a patient’s tumor is HRD-positive matters a lot because, once again, it can be exploited by a therapeutic! If a tumor is HRD-positive, regardless of whether the deficiency came from a BRCA1 deletion, a RAD51C mutation, or promoter methylation, it is often extremely sensitive to a class of drugs called PARP inhibitors.
So, understanding if a patient actually has HRD is both difficult and valuable. To help out with this, a company called Myriad Genetics developed myChoice, a test that computes a measure of HRD via a “genomic instability score” by integrating three measures of chromosomal damage: loss of heterozygosity, telomeric allelic imbalance, and large-scale state transitions, all extracted from the tumor. As far as I can tell from the technical documentation, the raw score itself, unlike gene signatures, has no intrinsic biological meaning. Its clinical utility comes entirely from an empirically determined threshold, established through population-level studies, that designates tumors as HRD-positive.
Mechanistically, we “know” that whatever the output of the myChoice algorithm is about DNA repair failure, but the exact construction of it is an empirical fit, not a first-principles derivation. Still, it works well enough for the FDA to have approved it as a companion diagnostic in 2021. Of course, the obvious question remains: is this black-box biomarker any better than human-legible ones? The answer does seem to be a tentative yes: 19%-61% patients identified as HRD-positive by the myChoice test would’ve been missed through simpler methods.
But even this test is white-box in the sense of the inputs (DNA measurements) to the model being legibly tied to the output (HRD-positive) of interest. In the most platonic form of ‘leaving things to the machine’, we would simply feed high-dimensional data to a model, and let it come to its own understanding—unabated by what humans think—of what is most important. For a very long time, this didn’t seem like a realistic clinical path forward, because purely data-driven biomarkers are hard to trust, hard to standardize, and hard to regulate. Yes, eventually the FDA would come around, but not anytime soon.
Yet in August 2025, for the first time ever, the cancer field saw the emergence of an FDA-authorized prognostic test that was exactly that: the ArteraAI Prostate Test.
All the test requires is a pathology slide (an ordinary H&E biopsy, the kind already produced for every prostate cancer patient) and a few standard clinical variables. A machine-learning model ingests those slides whole, millions of pixels at a time, and looks for patterns in the tissue architecture that no pathologist has ever consistently been able to describe. The model has no conception of “cells” or “glands,” but through training, implicitly learns the entire language of cellular morphology: the spacing of nuclei, the texture of stroma, the presence of inflammatory niches, and so on.
From this, it outputs two numbers: a risk score for 10-year metastasis rate, and, if the risk is high, a recommendation on whether the patient would benefit from abiraterone, a hormone therapy that reduces testosterone, starving prostate tumor cells. Most curious of all is that the basis of the approval hinged heavily on the model being applied to multiple prior Phase III trials across thousands of patients, demonstrating that the model could retrospectively predict which prostate cancer patient responded to hormone therapy.
This may be boring to pure machine-learning people. After all, the underlying model is, as far as I can tell from their initial paper, just a basic ResNet-50. But to people in the biotech space, this announcement is nothing short of insane. In fact, multiple parts of this are insane. Not only did the FDA approve a biomarker that was an entirely black-box readout with no human-legible intermediate criteria, they did so on the basis of an extremely large retrospective analysis. It is difficult to express how unexpected this is. Nearly every previous cancer biomarker that has ever made it into the clinic in the last 40 years has been validated prospectively, built into the design of a trial from the ground up, costing millions of dollars. Retrospective analyses in this field are typically hypothesis-generating, suggestive at best, and never enough to stand on their own. But here, it was enough for the FDA.
This should tell us two things.
One, our previous belief that many clinically useful variables are hiding within cancer datasets is almost certainly correct. Each of these variables are likely only weakly predictive when alone, but, if aggregated together, is enough to meaningfully stratify outcome. This is not a new hope; over the past decade, countless groups have trained neural networks on pathology slides, promising that “hidden morphologic signatures” could predict everything from molecular subtype to patient survival. How did ArteraAI succeed where others didn’t? Unfortunately, we do not know the answer, but it may come down to the same reason any given machine-learning tool succeeds where others failed: they simply executed better. Even if we agree that cancer is complex enough that machine intelligence is necessary to understand it, the rules of how to do that well remain tricky; slide-level heterogeneity, site-to-site variation in staining, and picking the wrong indication all still matter, and can sink an R&D effort if done incorrectly.
And two, the FDA is willing to accept biomarkers that are not directly tied to human-legible biological phenomena. Many people likely assumed that this would eventually happen, but few, including me, would’ve predicted that it could’ve possibly come as early as it did. But it has, and, more importantly, it does not seem like this is an edge case, but rather the beginnings of something new. Consider that in February 2025, the biotech startup onc.ai secured an FDA Breakthrough Device Designation for its ‘Serial CTRS’ system, which applies deep learning to CT scans to stratify non–small-cell lung cancer patients into high- and low-risk categories. Just like ArteraAI, their model does not use single, legible features such as lesion diameters, only the aggregated, weak latent patterns that their model has learned across the many CT scans in its training dataset.
So, what does the future hold?
Again, cancer has a surprising amount of detail, and it is unlikely that pathology images are alone able to explain everything about it. We have some empirical proof for this. A 2022 Cell paper compared how well a model performs across 14 cancer-outcome prediction tasks if given only pathology data, only molecular profile data (RNA, gene mutations, copy-number variation of the tumor), or both. The combined data won most of the time. A more recent 2024 paper from AstraZeneca says something similar, with the advantages of multimodality seeming to increase as the underlying datapoints grow in number.
To me, this implies that the the spoils of the cancer-understanding race will accrue to those who gather not just pathology, not just genomics, not just proteins, not just transcripts, not just epigenomics, not just plasma, not just the scientific literature, but all of them at once and more, fused into a single representation, and presented on a platter to an impossibly large statistical model for it to gorge itself on. What could such a model teach us? What about cancer has eluded centuries of human study upon it? What will ultimately require machine intelligence to make clear? The race is on to find out.
Afterword: I should mention that I work at Noetik, where we’re building multimodal foundation models of tumor microenvironments in order to predict response to cancer drugs. This essay grew out of countless conversations with colleagues about why cancer response prediction is so hard, and what will be necessary to improve it.